-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Advances in Life Sciences
p-ISSN: 2163-1387 e-ISSN: 2163-1395
2014; 4(5): 220-226
doi:10.5923/j.als.20140405.02
Pretreatment with Pyridostigmine Bromide Does not Induce Cellular Toxicity
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLSudeshna Bhattacharya1, Soma Haldar1, Sujata Roy1, Biplab Giri1, Shyamal Das Gupta1, Rahul Bhattacharya2, Pratiti Ghosh1
1Department of Physiology, West Bengal State University
2Division of Pharmacology & Toxicology, Defence Research and Development Establishment, Gwalior
Correspondence to: Pratiti Ghosh, Department of Physiology, West Bengal State University.
| Email: |  |
Copyright © 2014 Scientific & Academic Publishing. All Rights Reserved.
Soldiers are exposed to multiple stress conditions adverse psychological, physico-chemical and environmental conditions during warfare which can result in significant physical and chemical alterations in the biological system. The effects of carbamate (pyridostigmine bromide, PB) pretreatment prior to physiological stress and organophosphorous compound, DFP (diisopropylfluorophosphate) exposure have been investigated on rats. This study attempts to decipher the level of cellular toxicity imparted by PB pretreatment by assessing drug efflux transportation of the PB or DFP and possible genotoxicity with respect to chromosomal aberrations, in addition to total antioxidant status (TAS) of blood under such circumstances. Total antioxidant status was observed to be more than 50% potentiated with sign-free dose of PB. Immunohistochemical studies show that drug efflux transporter P-glycoprotein receptors were not quantitatively upregulated by physical stress or hyperthermia or hypothermia or sign-free dosage (0.075 mg/Kg after intramuscular injection) of the xenobiotic pyridostigmine bromide. Treatment with high doses (8-16mg/ml) of PB on alternate days for a week caused 10 -20% increase in the receptor count, indicating the toxicity level. Further, sign-free dose of pyridostigmine does not induce genotoxicity as far as chromosomal breakage is concerned. In silico docking studies show that the pyridostigmine molecule binds with maximum affinity to asparagine(1235), threonine(1199) and arginine(1229) in the C-terminal half of the P-glycoprotein. So, pretreatment with sign-free dose of pyridostigmine bromide offers sufficient protection from stress against organophosphorous DFP exposure as observed from these molecular studies and does not significantly alterP-glycoprotein receptors quantitatively.
Keywords: Pretreatment, Pyridostigmine Bromide, Cellular Toxicity
Cite this paper: Sudeshna Bhattacharya, Soma Haldar, Sujata Roy, Biplab Giri, Shyamal Das Gupta, Rahul Bhattacharya, Pratiti Ghosh, Pretreatment with Pyridostigmine Bromide Does not Induce Cellular Toxicity, Advances in Life Sciences, Vol. 4 No. 5, 2014, pp. 220-226. doi: 10.5923/j.als.20140405.02.
Article Outline
1. Introduction
- Organophosphorous (OP) compounds are used as pesticides and were also developed as chemical warfare agents. Pyridostigmine bromide (PB) is a quarternary ammonium compound which has been approved as a pretreatment drug against toxic OP compounds. A number of studies indicate that the drug was used to protect soldiers against nerve gas exposure during Persian Gulf War (PGW) but the veterans complained of various side effects ascribed to pyridostigmine although it was suggested as “symptom-free dose.” So, controversy hovers around the efficacy of PB with respect to its dosage. These symptoms are likely to be augmented by other stress factors like physical exercise in hyperthermic and hypothermic conditions [1]. Dose dependent efficacy of pyridostigmine pretreatment and time-dependent protection by physostigmine and pyridostigmine using maximum sign-free dose against inhaled sarin aerosols on rats have been reported [2].One of the key players affecting pharmacokinetic profiles of many clinically relevant compounds is an active efflux transporter, P-glycoprotein (Pgp) which is ubiquitously present in living systems from bacteria to humans. It is one of the most extensively studied transporters regarding drug resistance and drug-drug interactions. It is expressed predominantly at various physiological barriers and can influence drug absorption (intestinal epithelium, colon), drug elimination (kidney proximal tubules) and drug penetration at the blood-brain barrier (endothelial brain cells). So, P-glycoprotein mediated drug-drug interactions may occur at various organs and tissues of vertebrates and invertebrates and have been shown to interact and provide defense against environmental xenobiotics, including organophosphate pesticides [3]. P-glycoprotein also has a role in modifying insecticide toxicity in mosquitoes [4]. The Pgp inhibitor verapamil increased toxicity to three groups of insecticides, but not to the organophosphate chlorpyrifos. Structurally, P-glycoprotein is a membrane spanning protein, 1280 amino acids long, consisting of two homologous halves joined by a flexible linker region. Each half has the hydrophobic region containing six transmembrane domains followed by a hydrophilic domain containing a nucleotide binding site that can bind ATP and its analogues (Fig 1).
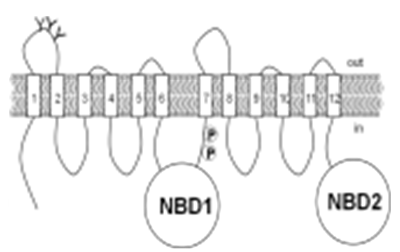 | Figure 1. 2-D Representation of P-glycoprotein |
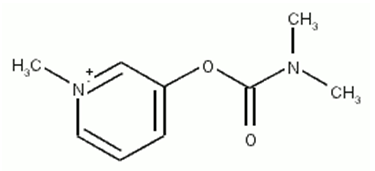 | Figure 2. Pyridostigmine molecule |
2. Methodology
2.1. Determination of LD50 of Pyridostigmine and Maximum Sign-Free Dose in Rats
- The LD50 of PB in male rats weighing 100±10g was determined by the method of Dixons Up and Down, for small samples. The maximum sign-free dose or symptom-free dose of pyridostigmine was evaluated by the method of Gordon et al, 1978 [12].
2.2. Measurement of Antioxidant Status
2.2.1. Treatment of Rats
- Pyridostigmine Bromide (PB) (Sigma Aldrich Co., Product # P797), was used for pretreatment against toxic organophosphorus compounds. A test group was treated with sign free dose (0.075mg/kg body weight) of PB on alternate days and another with higher dose (0.094mg/kg body weight) of PB for 5 consecutive days.
2.2.2. Estimation of Total Antioxidant Status
- Blood was collected from male Swiss albino rats (100+/-20gms) by decapitation after PB administration and assayed spectrophotometrically. (Callbiochem TAS assay kit, Catalogue No. 615700).
2.3. Immunohistochemical (IHC) Staining of P-glycoprotein Receptors
2.3.1. Treatment
- In one group, rats were pretreated with sign-free dose of PB and made to swim for 180 mins. This regimen was continued for 5 alternative days and the rats were sacrificed on the 11th day to make histological sections of intestine, liver and kidney. In the second group, pyridostigmine bromide at 0.075mg/kg body weight (intramuscularly) was administered for 7 alternate days and sacrificed after 7 more days. In the third group, the rats were given PB (0.075mg/kg body weight (intramuscularly) and sacrificed the next day for IHC studies. The fourth group was made to exercise for 3hours for 5 days and sacrificed on the sixth day; whereas the fifth group were subjects of hyperthermic studies and were also quantitated for P-glycoprotein.All experiments were conducted in triplicates.
2.3.2. Immunohistochemical Staining
- P-glycoprotein antibody [JSB-1] (ab3366) was used as the primary monoclonal antibody to detect the P-glycoprotein receptors immunologically [13, 14] according to the procedure of Abcam Co. The anti-ATP-binding cassette subfamily B (MDR/TAP), member 1A polyclonal antibody was bought from Millipore (catalogue no. AB10341). This recombinant ABC was presented with raw rabbit serum with 0.05% sodium azide and stored at -20°C. Paraffin sections of intestine, liver and kidney tissue of treated and control group of rats were deparaffinized with xylene, washed with 100% and 70% alcohol. Antigen was retrieved from tissue with phosphate buffered saline (PBS), washed with IHC buffer and incubated with blocking agent for 45 minutes. The primary antigen was added to it (1:150) for two hours and washed with 1X buffer to add secondary antigen for 20 minutes and rewashed with buffer to add streptavidin. DAB A and DAB B were mixed in the ratio of 1:25 and added to the tissues in dark condition and incubated for 10mins for color development. Subsequently it was stained with hematoxyline and mounted for viewing.
2.4. Chromosomal Aberration
- The control group of rats were treated with colcemid (colchicine) which acts as a potent cell cycle inhibitor. The method of chromosomal staining was followed according to Guo et al (2008) [15].
2.5. Pyridostigmine-P-glycoprotein Interaction in silico
- This method provides a qualitative, quick and inexpensive way of evaluating potential drug interaction. Computational structure prediction of ligand-protein complexes may be performed using docking methods like Auto-dock vina, DOCK, FLEXx and GOLD [16] in combination with ADT tools. This will help predict ligand orientations in binding sites and binding affinities of ligands.Pyridostigmine-P-glycoprotein interaction has been assessed using Auto-dock vina. Docking was performed with the AutoDock program (version 4.00) [17]. Vina input file configuration was used. Eight independent docking runs were carried out for the ligand pyridostigmine, starting from randomly generated initial conformations of the ligand. We used the genetic algorithm in AutoDock to perform the global search, completed with a local search, as it has been shown that it gives efficient sampling. The binding zone was a cube whose length was set to 30 Å, at the surface exposed to the exterior of the cell membrane. The rate of crossover was 80% and the mutation rate was fixed to 2%. Such analyses were performed using the N-terminal and C-terminal halves of Pgp using ADT tools to prepare the ligands and receptors for Auto Dock vina. RASMOL Version 2.6 was used as viewer. The drawback of the docking was that hydrogens could not be assigned to the protein.
3. Results
3.1. Determination of LD50 of Pyridostigmine and Maximum Sign-Free Dose in Rats
- LD50 of Pyridostigmine bromide was found to be 1.88 mg/Kg after intramuscular injection.The maximum symptom-free dose of pyridostigmine which caused no sign of anticholinesterase poisoning such as tremors, muscle fasciculation, salivation, urination, incoordination, was found to be 0.075 mg/Kg after intramuscular injection.
3.2. Measurement of Antioxidant Status
- Total antioxidant status evaluation showed that with sign-free dose, the antioxidation level of PB-treated (0.075mg/kg) rats was significantly increased at 0.113 level, being 51.68% more potentiated as compared with the control group. (Fig. 3)
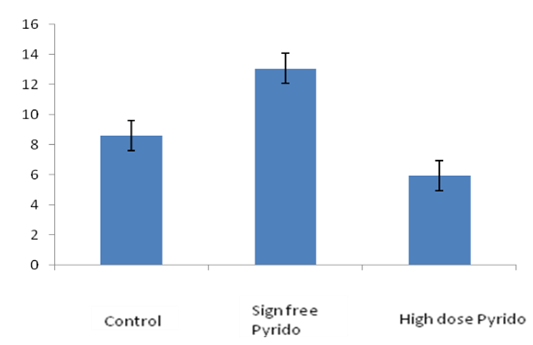 | Figure 3. Total antioxidant status increases with sign-free dose of pyridostigmine and decreases with higher dosage |
3.3. Quantification of P-glycoprotein Receptor: Immunohistochemical (IHC) Staining of P-glycoprotein Receptors
- Immunohistochemical studies show that there was no increase in P-glycoprotein receptors with 0.075mg/kg bw dose of pyridostigmine compared to the control sets but there was 50–100 fold increase with 10–16mg PB/kg bw/day. Animals sacrificed immediately after high dose administration could not show any histological change (Fig. 4).
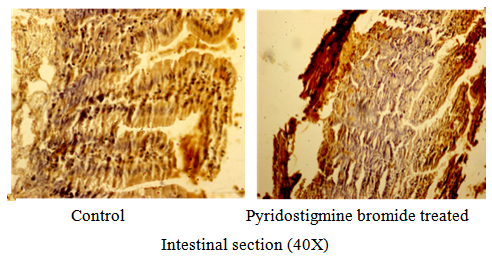 | Figure 4. There was no change in the quantity of P-glycoprotein receptors with sign-free dose of Pyridostigmine Bromide |
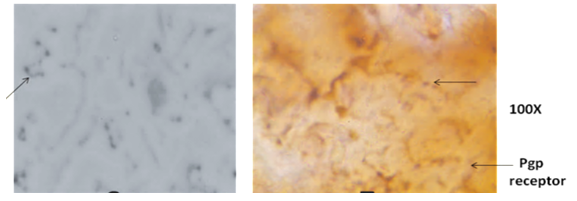 | Figure 5. Liver section showing increased P-glycoprotein receptor levels by 50 – 100 fold with 10 – 16mg PB /kg/day |
3.4. Chromosomal Aberration with PB Administration
- Dicentric chromosomes are reliable indicators of chemical exposure. Cytogenetic study of PB pretreated rats was used for preparation of metaphase chromosomes which is frequently used to reveal chromatid aberration.Present study with sign free dose of pyridostigmine bromide (0.075 mg/kg body weight) administration revealed no chromosomal aberration, despite dose increment to 0.094 mg/body weight (Fig.6). So, pyridostigmine does not induce genotoxicity as far as chromosomal breakage is concerned, with the existing sign-free dosage regimen.
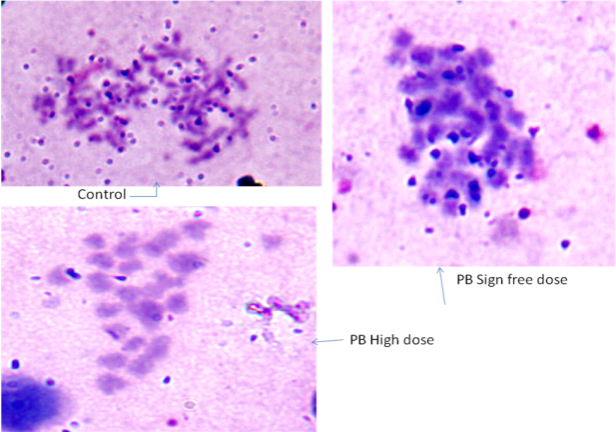 | Figure 6. No chromatid aberration was observed with pyridostigmine bromide administration at sign-free dose or high dose |
3.5. Pyridostigmine-P-glycoprotein Interaction in silico
- In this study a computational approach allows prediction of whether pyridostigmine is likely to interact with P-glycoprotein. The mouse P-gp crystal structure revealed a large and hydrophobic binding cavity with no clearly defined sub-sites that supports an ‘‘induced-fit’’ ligand binding model. Flexible receptor docking was employed to develop a new prediction algorithm for P-gp binding specificity. Many P-gp substrates bind deeper in the cavity and specificity in P-gp is better understood in terms of physicochemical properties of the ligands (and the binding site), rather than specific sub-sites. Interestingly all docked poses for each ligand are found to interact with residues which have been experimentally identified to bind a specific ligand. The docked poses for pyridostigmine roughly sample two locations, size and distance between the binding site residues [18, 19].The pyridostigmine molecule binds with maximum affinity (energy value = -4.0 Kcal/mole), with asparagine (1235), threonine(1199) and arginine(1229) in the C-terminal half of the protein, exposed to the interior surface of the cell (Fig.7).
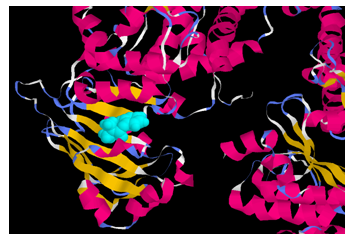 | Figure 7. Docking studies show that the pyridostigmine molecule (in blue) binds with maximum affinity in the C-terminal half of the P-glycoprotein |
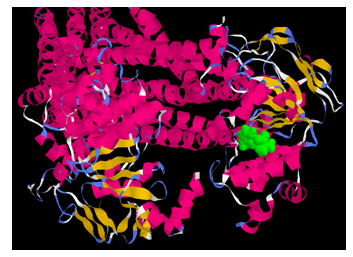 | Figure 8. Energy value of binding of pyridostigmine (green) in the N-terminal arm with GLN (625), VAL (622) and ARG (584) is -3.7 Kcal/mole |
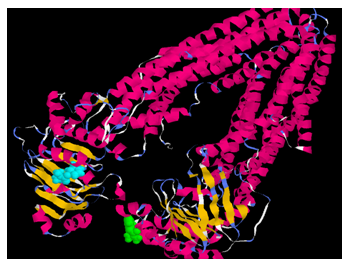 | Figure 9. Pyridostigmine molecule (blue in C-terminal, green in N terminal) binds with maximum affinity with asparagine(1235), threonine (1199) and arginine(1229) in the C-terminal of the P-glycoproteinmolecule |
5. Discussion
- The LD50 value and sign-free dosesconcur with earlier reported values [20].Antioxidant defenses are essential for cellular redox regulation. Since free-radical production may be enhanced by toxicants, the effect of PB on total antioxidant status (TAS) was evaluated to find that total antioxidant status rises with minor dose of PB and offers proper redox balance at the this dose but not so at the higher dose. It is possible that the entire system could be geared up for resetting of homeostasis and the maximum capacity for detoxification was in action and thus the challenge with the minute drug amount offered protection. Somani et al (2000), showed that physical stress enhanced the delayed toxic effects of a subchronic oral dose of pyridostigmine primarily in the skeletal muscle of mice [21]. Cytogenetic damage induced by sign-free dose and slightly higher dose of PB injected intraperitoneally into rats was evaluated by determining the frequencies of chromosomal aberrations. No significant aberration, breaks, gaps or polyploids were observed with the injected doses. This indicates that slight increase in the pretreatment dose may not be harmful against toxic OP compounds. Such studies have been done with fenvalerate [22] in microorganisms with conflicting results and with uranyl fluoride [23] in mice.P-glycoprotein is an ABC transporter, a member of the ATP Binding Cassette group of membrane spanning transporters, whose physiological function is to protect against toxicological (chemical) stress [24]. It plays a vital role in effluxing a wide range of xenobiotics through gastro-intestinal tract, kidney, liver and blood brain barrier. Multiple drugs against cancer, HIV [25] etc. serve as its substrate which may be circumvented by modulators [26] or inhibitors. P-glycoprotein is regulated by physical and psychological stress as its expression is regulated by osmotic stress [27] and heat-shock transcription factor-1 [28]. Chronic stress and antidepressant treatment have opposite effects on P-glycoprotein at the blood–brain barrier [29]. PB is a reversible cholinesterase inhibitor which does not cross the blood brain barrier (BBB) except in stressful conditions.Pyridostigmine and diisopropylfluorophosphate (DFP) are known toxicants, so it is possible that P-glycoprotein has a role in their efflux transportation, in which case it should be overtly manifested on the cell, which can be detected immunohistochemically by quantification of the increased number of the P-glycoprotein receptors. As treatment with DFP was for a very short time span and receptor upregulation is a genetic process requiring an induction period, as expected, it failed to cause any change in receptor quantity. As the pre-existing sign-free dose did not cause any increase in the receptor quantity with acute or chronic exposure to PB, the dose may be regarded as nontoxic. Transport studies of this ATP Binding Cassette (ABC) transporter, which regulates the CNS drug homeostasis, can confirm the interaction of carbamates with P-gp. The observed interaction is sufficient to cause transient conformational change in the P-glycoprotein transporter resulting in energy liberation for drug efflux from the interior of the cell.
6. Conclusions
- The present study shows that the pretreatment dose of PB is non-toxic at the cellular level. The total antioxidant status as reflected in the serum of rats shows that the existing sign-free dose of PB confers sufficient protection. Immunohistochemical studies reveal that sign-free dose of pyridostigmine bromide does not quantitatively upregulate efflux transporter receptors. Treatment with higher doses causes 10-20% increase in the receptor count, indicating the toxicity level. Physical stress or temperature deviations do not show any significant change. Also, pyridostigmine does not induce genotoxicity with the existing sign-free dose as far as chromosomal breakage is concerned.Docking studies have shown that the pyridostigmine molecule binds with maximum affinity with asparagine (1235), threonine (1199) and arginine (1229) in the C-terminal end of the P-glycoprotein. This shows that the drug is capable of being bound to the receptor as its substrate and thus be effluxed out by it according to environmental demand and the degree of toxicity imposed on the system. Binding does not necessarily infer transport, so efflux assay of this ATP Binding Cassette transporter, with these drugs, need to be performed to confirm the interaction of carbamates with P-glycoprotein.
ACKNOWLEDGEMENTS
- This project was funded by DRDE Gwalior, vide PROJECT NO: SS -10 / DRDE / NBCD / S&T-28/01.