-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Materials and Chemistry
p-ISSN: 2166-5346 e-ISSN: 2166-5354
2013; 3(A): 1-7
doi:10.5923/s.ijmc.201303.01
In Vitro Bioactivity and MG63 Cytocompatibility of Chitosan-Silicate Hybrids
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLYuki Shirosaki1, Tomoyuki Okayama2, Kanji Tsuru3, Satoshi Hayakawa2, Akiyoshi Osaka2
1Frontier Research Academy for Young Researchers, Hibikino, Wakamatsu-ku, Kitakyushu, 8080196, Japan
2Graduate School of Natural Science and Technology, Okayama University, Tsushima, Kita-ku, Okayama, 7008530, Japan
3Faculty of Dental Science, Kyushu University, Maidashi, Higashi-ku, Fukuoka, 8128582, Japan
Correspondence to: Yuki Shirosaki, Frontier Research Academy for Young Researchers, Hibikino, Wakamatsu-ku, Kitakyushu, 8080196, Japan.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
This paper describes in vitro bioactivity and cytocompatibility of chitosan-silicate hybrids derived from chitosanand γ-glycidoxypropyltrimethoxysilane (GPTMS) through sol-gel processing. Chitosan, one of the biodegradable polymers, was selected as a hybrid matrix whereas GPTMS was employed as crosslinking agent as well as bioactive inorganic species. GPTMS was hydrolyzed completely in acetic-acid solution to yield Si-OH groups, which in turn were condensed to form Si-O-Si networks. Cytotoxicity of GPTMS monomer was much lower than that of glutaraldehyde. From MG63 osteoblastic cell culture, the chitosan-GPTMS hybrids were cytocompatible better than the hybrids crosslinked with glutaraldehyde, tetraethoxysilane or glycidylmethaacrylate. The hybrids incorporated with Ca ions deposited apatite on their surface under a simulated body fluid (SBF, Kokubo solution). Freeze-drying lead to porous hybrids with about 90% in porosity and about 100 μm in size. When soaked in SBF, the porous deposited apatite not only on the outer surface but also on the pore wall. The porous hybrids hence are considered to be appropriate materials for scaffolds.
Keywords: Chitosan, γ-glycidoxypropyltrimethoxysilane (GPTMS), Organic-Inorganic Hybrid, Scaffold, Cytocompatibility, Apatite, Simulated Body Fluid
Cite this paper: Yuki Shirosaki, Tomoyuki Okayama, Kanji Tsuru, Satoshi Hayakawa, Akiyoshi Osaka, In Vitro Bioactivity and MG63 Cytocompatibility of Chitosan-Silicate Hybrids, International Journal of Materials and Chemistry, Vol. 3 No. A, 2013, pp. 1-7. doi: 10.5923/s.ijmc.201303.01.
Article Outline
1. Introduction
- The success of tissue engineering primarily depends on the scaffolds that serve to support and reinforce theregenerating tissue. The scaffolds should not only promote cell adhesion, cell proliferation, and cell differentiation, but also should be biocompatible, biodegradable, and highly porous[1]. Chitosan consists of polysaccharide chains, and is one of the candidate materials for scaffolds since they are bioresorbable as well as biocompatible, non-antigenic and non-toxic[2]. Yet their inferiority in mechanical properties and too high in vivo biodegradability are disadvantageous for most medical applications. Hence, mechanical strength, and biodegradation are to be controlled within proper allowance or an acceptable range before chitosan is employed for scaffolds. For such purposes, cross-linking thepolysaccharide chains with a non-toxic agent to form a networked is essential. Various cross-linking agents have been attempted such as epoxy compounds and aldehydes[3, 4], whereas they are all highly cytotoxic and may spoil the biocompatibility of chitosan. Shirosaki et al.[5] have already pointed out thatγ-glycidoxypropyltrimethoxysilane (GPTMS) is a good cross-linking agent for chitosan, and that the chitosan- GPTMS hybrids are far better than chitosan incytocompatibility with human osteosarcoma cell MG63. Moreover, they might induce apatite nucleation and growth under the body environment or in a simulated body fluid (Kokubo solution,) since they involve inorganic species (silanol groups, Ca2+) that are considered essential for inducing the apatitenucleation[6]. Such ability of spontaneous apatite deposition is often denoted as bioactivity. If they are bioactive, they might be fixed in tissue defects with forming direct bonds. That is, they are employable as scaffolds, tissue substitutes, or filler for tissue defects. In this study, we examined in vitrobioactivity and cytocompatibility of chitosan-GPTMS hybrids as well as the structure of their hybrids. Moreover, freeze - drying technique is used to prepare porous chitosan -GPTMS hybrids and their applicability to tissue engineering scaffolds is explored.
2. Materials and Methods
2.1. Preparation of the Hybrids
- An appropriate amount of chitosan (high molecular weight, deacetylation: 79.0%, Aldrich®, USA) was dissolved in 0.25M acetic acid aqueous solution to attain a concentration of 2% (w/v), to which γ-glycidoxypropyltrimethoxysilane (GPTMS; Chisso, Tokyo, Japan,CH2OCHCH2OCH2CH2Si (OCH3)3) and calcium chloride (CaCl2, Nacalai Tesque, Kyoto, Japan) were added so that the solutions with compositions in Table 1 were obtained. Here, 1mole chitosan stands for 1 mole deacetylated amino groups of their unit. Table 1 also listed the samples codes. Tetraethoxysilane (TEOS), glutaraldehyde (GA), and glycidylmethacrylate (GMA) were used as another crosslinking reagents for comparison. After stirring at room temperature for 1h, the resultant solutions were poured into polypropylenecontainers. The resultant solutions were aged in the shielded polypropylene containers at 60°C for 2 days. Then, the gel-like hybrids were dried at 60°C for 2 days to yield hybrid membranes. The obtained membranes were soaked in 0.25N sodium hydroxide to neutralize and wash out the remaining acetic acid, and rinsed well with distilled water. In addition, freeze-drying was used to derive porous hybrids: A fraction of each resultant sol was poured into a polystyrene container, and frozen at -20°C for 24h in the refrigerator before vacuum drying.
|
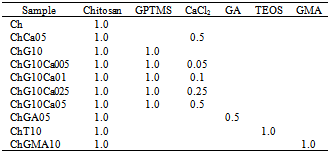
2.2. Structural Characterization of the Hybrids
- Fourier-transform infrared (FT-IR) spectra were measured with a FT-IR spectrometer (Nexus470, Thermo Nicolet) using the KBr method, where the signals from 128 scans were accumulated with a resolution of 4 cm-1. The pore diameter was evaluated with the optical microscope images. Outlines of the pore were traced and the maximum diameter was derived using Image-Pro Plus software (Planetron, Tokyo, Japan). In each procedure, at least twenty pores were picked up from three different areas of a sample. The density of the scaffolds was derived from the weight and geometric volume.
2.3. Cell Culture
2.3.1. Cytotoxicity of the Cross-linking Agents
- On degradation under the body environment, the hybrids may release several fragments into plasma. Since, chitosan fragments have been proved non-toxic. It should be the first thing to examine cytotoxicity of debris from cross-linking reagents: it was evaluated in vitro using osteoblastic cells MG63 cells (DAINIPPON PHARMACEUTICAL Co., LTD, Japan). MG63 cells were seeded in 96-well plates at 6400 cells per well (2x104 cells·cm-2) in 250 μl D-MEM with 10% FBS, 1 % penicillin and streptomycin solution and 2.5 μgml-1 fungizone and supplemented with 50 μgml-1 ascorbic acid, 1% MEM-non essential amino acids and 2.0 mMml-1 L-glutamine. GA and GPTMS were added to the culturing media up to 100 ppm and 1000 ppm, respectively. The cell culture system was maintained in a humidified incubator at 36.5°C with 5% CO2 in air. After 24h of culture, surviving cell numbers were determined indirectly by MTT assay. MTT50 represents the concentration of each cross-linking agent where the absorbance were half that of the cells without agent.
2.3.2. Cytocompatibility of the Solid Membrane
- The solid membranes were soaked in PBS (pH7.4) solution and sterilized by autoclaving at 121°C for 20 min. MG63 cells were cultured (104 cells·cm-2) under control conditions (absence of materials, standard plastic culture plates) and on the surface of the solid membranes. The cultures were evaluated throughout the incubation time at days 1,3, 5, and 7 for cell viability and observed by scanning electron microscopy (SEM). The cell viability was determined by MTT assay.
2.4. Evaluation of in Vitro Apatite Formation
- SBF was prepared as described in the literatures[6]. It has been proved that in vivo apatite formation on bioactive implants is almost fully reproduced under in vitro experiments in SBF. The solid membranes (30x10x1mm2) and porous scaffolds were soaked in 30 ml SBF held in capped polystyrene bottles at 36.5°C up to 14 days as pH was monitored. Then, they were gently rinsed with distilled water and dried at room temperature. Their surfaces were characterized using an X-ray diffractometer with a thin-film measuring attachment (TF-XRD): RAD-II AX, CuKα, (Rigaku, Tokyo, Japan), operated at 40kV-20mAacceleration. The surface morphology was observed under a scanning electron microscope (SEM: JEOL, JMSM-6300, Tokyo, Japan) equipped with an energy-dispersion X-ray (EDX) analyzer (model DX-4, Phillips). The concentrations of the component elements released into SBF were measured by inductively coupled plasma emission spectroscopy (ICP; ICPS-7500, Shimazu, Japan).
3. Results and Discussion
3.1. The Solid Membrane
- Fig. 1 shows the FT-IR absorption spectra of thespecimens. The bands at 1650, 1565, and 1110-1000 cm-1 were assigned to the constituent bonds of chitosan and GPTMS[5]. Growth of a component (Si-O) in the band at 1100-1000 cm-1 confirmed the formation of Si-O-Si bridging bonds on hybridization[5]. Difference in IR profile was detected between ChG10Ca05 and ChG10 in the peaks assigned to the amino groups. This should be attributed to the presence of Ca(II) in the system, suggesting that the chemical environment around the amino groups is different among the solid membranes with an without Ca(II). Nishi et al.[7] suggested the presence of good interactions between the amino groups of chitosan and Ca2+ to form complexes. Thus, though no more define data are available at this moment, one might suppose the presence of any calcium-amino group interactions.
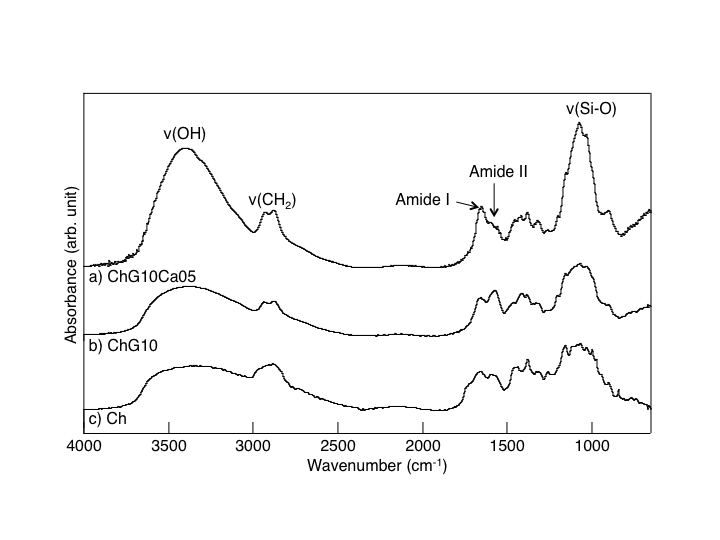 | Figure 1. FT-IR absorption spectra of (a) ChG10Ca05, (b) ChG10, and (c) Ch |
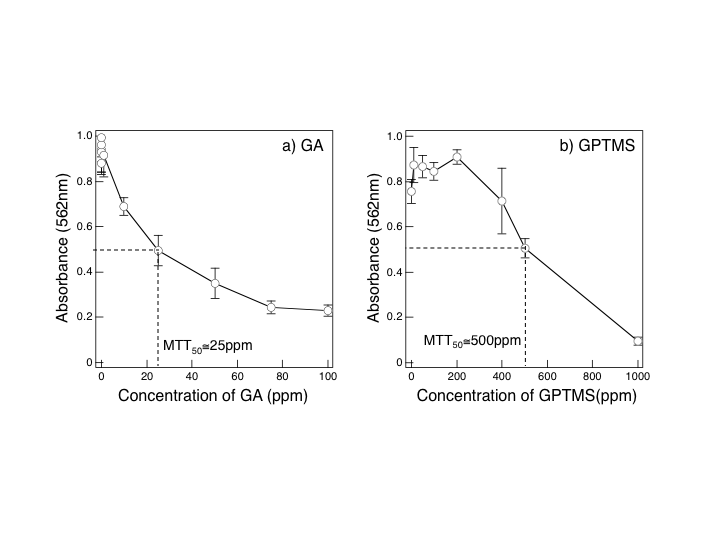 | Figure 2. MTT assay for MG63 cells cultured in the media added with GA (a) and GPTMS (b): ~25 and ~500 ppm for MTT50, respectively. See text for MTT50 |
 | Figure 3. Morphology of MG63 cells cultured for 1 day on the solid membranes (a) Ch without cross-linking agent, and cross-linked with (b) GPTMS(ChG10), (c) GA(ChGA05), (d) GMA(ChGMA10), and (e) TEOS(ChT10), respectively |
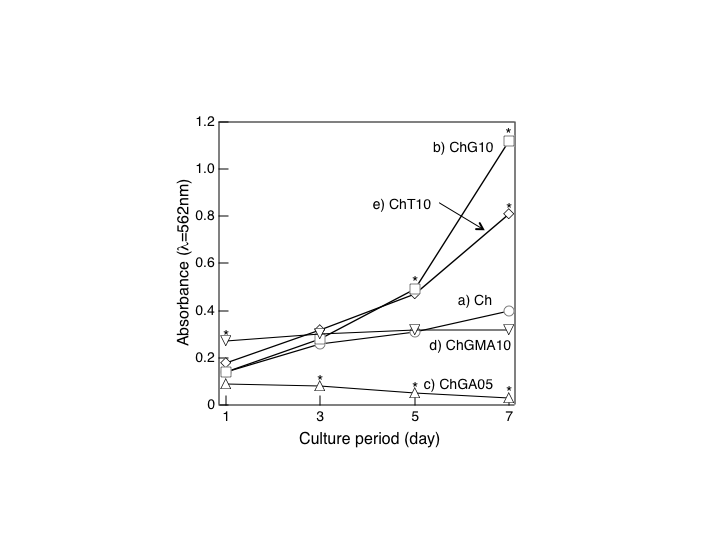 | Figure 4. Cell proliferation (MTTassay) of MG63 cells cultured on the solid membranes (a) Ch without cross-linking agent, and cross-linked with (b) GPTMS(ChG10), (c) GA(ChGA05), (d) GMA(ChGMA10), and (e) TEOS(ChT10), respectively |
 | Figure 5. Morphology of MG63 cells cultured for 7d on ChG10 (left) and ChT10 (right) |
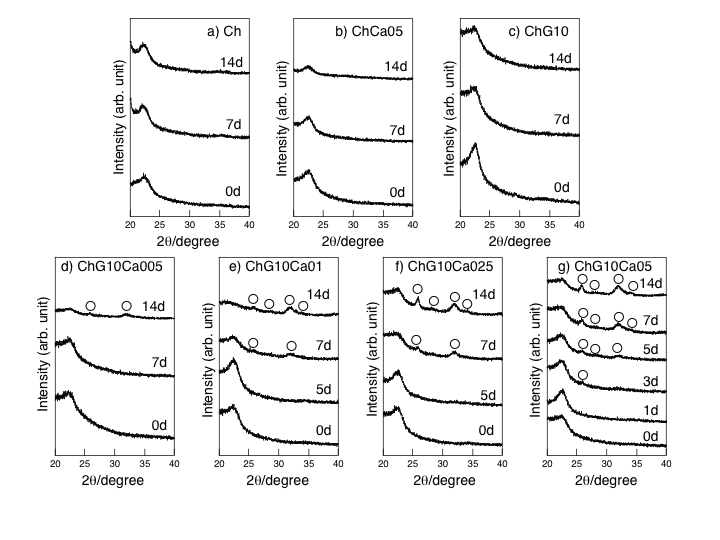 | Figure 6. TF-XRD patterns for the hybrid membranes before and after being soaked in SBF up to 14 days. (a) Ch, (b) ChCa05, (c) ChG10, (d) ChG10Ca005, (e) ChG10Ca01, (f) ChG10Ca025, and (g) ChG10Ca05. ○:apatite |
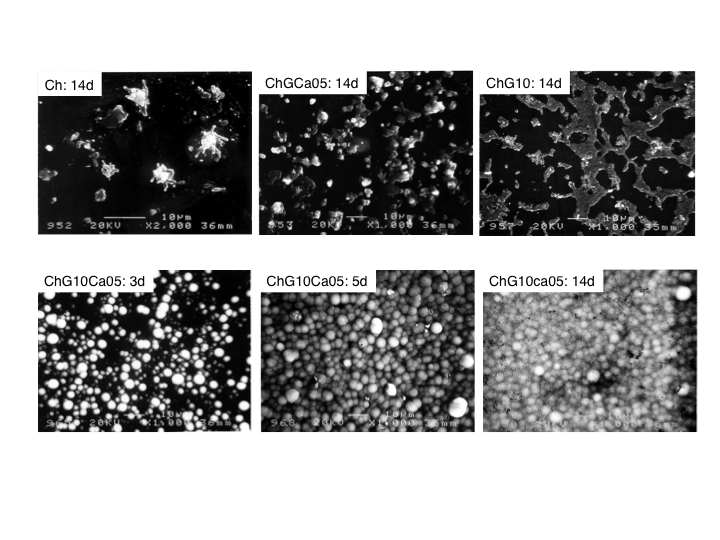 | Figure 7. SEM images of the surface microstructure of the solid membranes. Top row: Ch, ChCa05, and ChG10 soaked in SBF for 14d. Bottom row: ChG10Ca05 soaked in SBF for 3 to 14d. Bar : 10 μm |
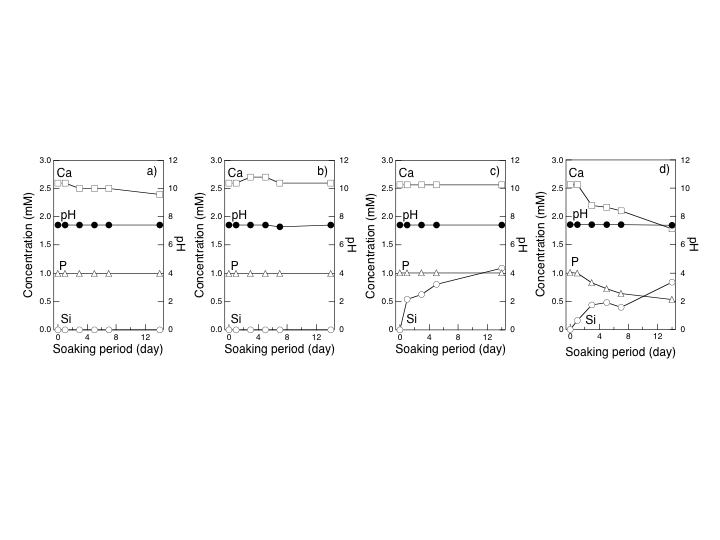 | Figure 8. The concentration of Ca(II), P(V), and Si(IV) and pH of SBF after soaking selected solid membranes: (a) Ch, (b) ChCa05, (c) ChG10, (d) ChG10Ca05 |
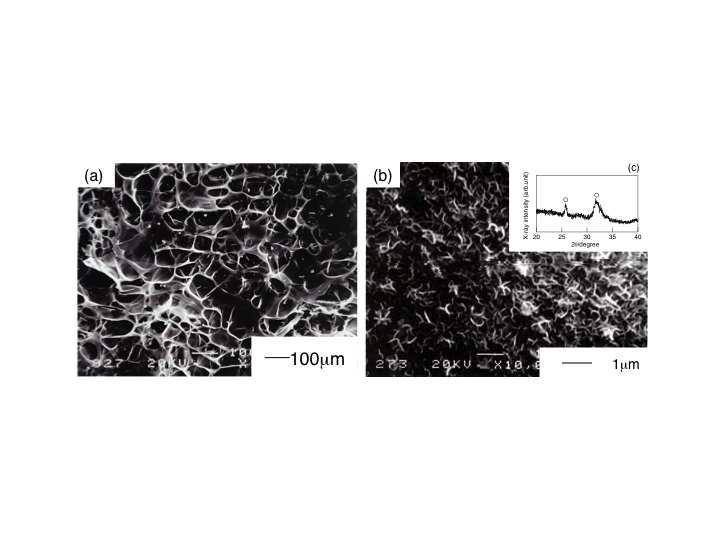 | Figure 9. SEM images of the as-prepared porous scaffold ChG10Ca05 (a), and (b) the scaffold after soaked in SBF for 7d. The XRD profile of insert (c) indicates the flakes of (b) are apatite crystallites |
3.1. The Porous Scaffolds
- Fig. 9(a) shows a SEM image of a porous hybrid, ChG10Ca05, as a typical porous microstructure of the chitosan-hybrid scaffolds obtained in the present study due to freeze-drying. The pore size and porosity of the scaffolds was approximately 100 μm and about 90%, respectively. Moreover, the pores were interconnected with windows of walls open to adjacent pores ~50 μm, large enough for supply oxygen and nourishment. Those pores were within the range to promote effective ingrowth of bone tissue and to provide mechanical interlock for firm initial fixation, proposed by Helebert et al.[12] and de Groot[13]. When soaked in SBF for 7d, the porous ChG10Ca05 scaffolds deposited apatite not only the outer surface but also on the pore walls. The inserted XRD profile confirmed that the flaky deposits were apatite. This concludes that the present porous scaffolds have good bioactivity as they maintained the original porous configuration and flexibility. Therefore, the porous scaffolds are promisingly applicable to bone tissue engineering scaffolds.
5. Conclusions
- The toxicity of GPTMS monomer was much lower than that of GA. MG63 osteoblastic cell cultured onchitosan-GPTMS solid membranes showed good cytocompatibility compared with the hybrids crosslinked by other reagents such as GA. Ability of the apatite deposition on the solid membranes was accelerated not only the release of Ca ions from the hybrids but also that of Si ions. Porous hybrids were easily prepared by freeze-drying technique and deposited apatite even the walls of the inside pores under the simulated body fluid. Therefore, it has been concluded that the chitosan-GPTMS porous hybrids are promisingcandidates of basic materials from which tissue engineering scaffolds are to be derived.