-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Biophysics
p-ISSN: 2168-4979 e-ISSN: 2168-4987
2013; 3(1A): 18-38
doi:10.5923/s.biophysics.201311.03
Single-molecule FRET Studies on DNA Mismatch Repair
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLJeungphill Hanne1, Jiaquan Liu1, Jong-Bong Lee1, 2, 3, Richard Fishel1, 4
1Department of Molecular Virology, Immunology and Medical Genetics, The Ohio State University Medical Center, Columbus, USA
2Department of Physics, Pohang University of Science and Technology (POSTECH), Pohang, Korea
3School of Interdisciplinary Bioscience and Bioengineering, POSTECH, Pohang, Korea
4Physics Department, The Ohio State University, Columbus, USA
Correspondence to: Richard Fishel, Department of Molecular Virology, Immunology and Medical Genetics, The Ohio State University Medical Center, Columbus, USA.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
DNA mismatch repair (MMR) is the process that corrects misincorporation errors introduced by DNA polymerases during replication. MMR is also associated with other biological processes such as the suppression of recombination between partially homologous sequences (homeologous recombination) and DNA-damage signaling. Mutations of the human MMR genes are the cause of Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer (LS/HNPCC). The detailed mechanism of MMR components in these biological processes remains enigmatic. MMR that is coupled to replication is an excision-resynthesis reaction. It is initiated at a distant strand scission that is 100’s to 1000’s of bp from the mismatch and the excision tract extends from that strand scission to just past the mismatch. MutS and MutL are unique core components of MMR that recognize a mismatch and initiate the excision reaction. Additional components include the replication clamp (β-clamp in prokaryotes and PCNA in eukaryotes), the replication clamp loader (γδδ’ in prokaryotes and RFC in eukaryotes), an exonuclease (Exo1, ExoX, RecJ, ExoVII in prokaryotes and EXOI in eukaryotes), and single strand binding protein (SSB in prokaryotes and RPA in eukaryotes). Recently single molecule FRET/Fluorescent Tracking (smFRET/FT) studies have made a significant impact on understanding the MMR process. More extensive future smFRET/FT studies are expected to further detail the MMR mechanism. This review is intended to offer a guide to applying smFRET/FT studies to understand the entire MMR process, by pinpointing key questions and poorly understood phenomena. We summarize recent smFRET/FT results and address major issues in the application of the smFRET/FT system.
Keywords: Total Internal Reflection Fluorescence, TIRF, ATPase, Exonuclease, Protein Interactions
Cite this paper: Jeungphill Hanne, Jiaquan Liu, Jong-Bong Lee, Richard Fishel, Single-molecule FRET Studies on DNA Mismatch Repair, International Journal of Biophysics , Vol. 3 No. 1A, 2013, pp. 18-38. doi: 10.5923/s.biophysics.201311.03.
Article Outline
1. Introduction
- The concept of mismatch repair (MMR) was independently proposed in 1964 by Evelyn Witkin based on her bromo-deoxyuracil incorporation studies[1], and Robin Holliday based on his gene conversion analysis in fungi[2]. In the early 1970s the genes responsible for MMR started to be identified[3,4]. Since then the mechanism of MMR has been continuously refined[5-8]. Components of the MMR machinery have been shown to be involved in other biological phenomena such as the suppression of recombination between partially homologous DNAs (homeologous recombination)[9], and DNA-damage signaling[10-12]. Defects in core human MMR genes are the cause of Lynch syndrome also known as hereditary nonpolyposis colorectal cancer (LS/HNPCC); perhaps the most common hereditary cancer predisposition syndrome [13-18]. In this section, we briefly summarize the basic MMR process and its role in other biological processes as well as point out key unanswered questions.
1.1. Replication Coupled DNA Mismatch Repair
- The basic role of MMR is to correct nucleotide misincorporation errors introduced by the DNA polymerase during DNA replication. Indeed, the MMR genes were originally identified as “mutators” (Mut) since uncorrected replication errors resulted in a 100-1000 fold increase in spontaneous mutation rates[19-21]. The core genes of MMR have been conserved through evolution (Table 1; Fig. 1)[22]. The MMR mechanism can be sequentially divided into four main steps: 1.) Recognition of a DNA mismatch, 2.) Transfer of the mismatch recognition signal to the distant strand scission site where DNA excision starts, 3.) Excision of the “newly incorporated” mismatched strand[5-8] and 4.) resynthesis of the excised DNA strand. Strand-specific excision engenders the unique steps in MMR since the resynthesis step appears to be accomplished by the normal replication polymerase machinery.
|
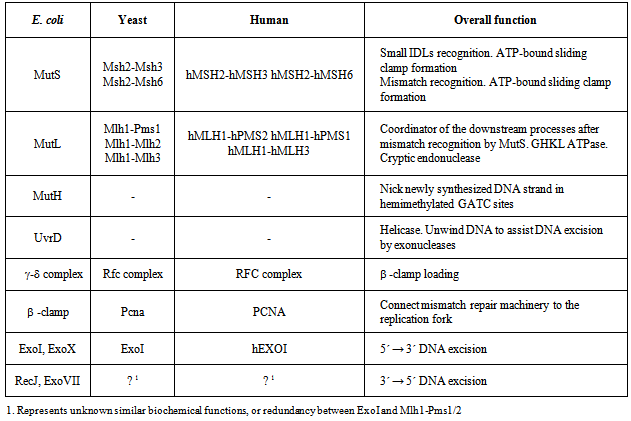
 | Figure 1. DNA Mismatch Repair in Prokaryote and Eukaryote Systems. A. Prokaryote MMR (gram negative enteric bacteria E.coli). MutS recognizes the mismatch and MutS and MutL together activate the MutH endonuclease. MutH introduces a strand scission on the unmethylated strand of a nearby hemi-methylated GATC site (-CH3). The newly replicated strand is transiently unmethylated following replication ultimately providing strand discrimination for MMR. The strand scission serves as an entry site for the UvrD helicase and one of four exonucleases that unwind and degrade the daughter strand to just past the mismatch. E.coli SSB protects the template single strand DNA until the replication machinery reengages to synthesize the complementary strand. The remaining strand scission is sealed by DNA ligase, completing the MMR process. B. Eukaryote MMR (and prokaryote MMR other than gram negative enteric bacteria). Mismatched nucleotides are primarily recognized by MSH2-MSH6. MSH2-MSH6, MLH1-PMS2 (Pms1 in S.cerevisae) and EXOI degrade the mismatched strand, starting at scissions on the newly synthesized strand. The single-stranded DNA binding heterotrimer RPA protects the ssDNA gap, while the replication machinery rengages to synthesize the complementary strand. Remaining strand scissions are sealed by DNA ligase |
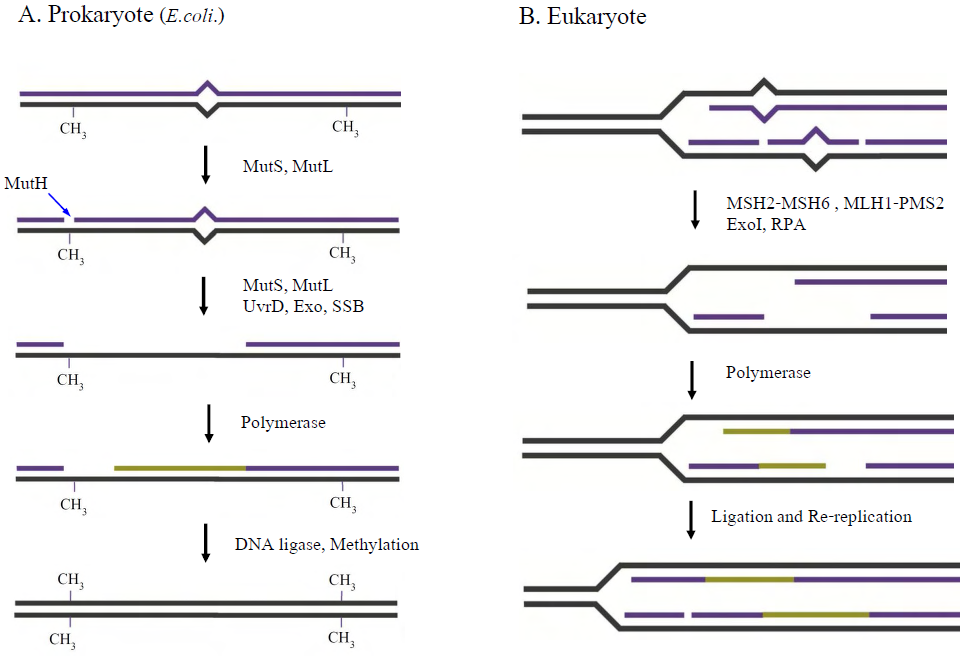
 | Figure 2. Models for the Mismatch Recognition and Downstream Signal Transfer. Left) Static Transactivation. The MSH or a complex of MSH and MLH/PMS capture a random loop of DNA to authorize the distant site where helicase/exonuclease may start degradation of the newly replicated strand. (Center) Hydrolysis-Dependent Translocation. ATP binding and hydrolysis by the MSH and MLH/PMS elicit movement to the distant target site where helicase/exonuclease may start degradation of the newly replicated strand. (Right) Molecular Switch Sliding Clamp. The mismatch-dependent ATP-binding provokes a conformational transition of MSH into a stable sliding clamp on the DNA that recruita MLH/PMS. Movement of MSH-MLH/PMS complex is driven by thermal diffusion that ultimately reaches to the distant excision-initiation site |
|
1.2. Suppression of Homologous Recombination by DNA Mismatch Repair
- MMR suppresses recombination between partially homologous sequences (homeologous recombination) [9,65,66]. The mechanism is unknown. Early studies suggested that MMR components inhibited the strand exchange reaction (recombinase) catalyzed by the central recombination-initiation protein RecA[67]. However, since both MutS and MutL may individually bind single-stranded DNA (ssDNA), it remains formally possible that the observed inhibition of RecA recombinase was a result of competition for the homologous donor ssDNA rather than discrimination between partial homology with the acceptor double-stranded DNA (dsDNA). Other possible mechanisms would include: 1.) recognition of mismatches in the D-loop recombination initiation intermediate that provokes MMR excision and ultimately release of the invading donor ssDNA, or 2.) MMR-dependent incision of the D-loop that would release the superhelicity required to stabilize the homologous donor ssDNA. It is also possible that aspects of both these models are operable.
1.3. Involvement of DNA Mismatch Repair in DNA-damage Signaling
- MMR deficient cells are at least 100-fold more tolerant to the exposure of various toxic agents that damage DNA. It has been suggested that MMR proteins act as damage sensors that initiate an apoptotic response following overwhelming or unrepairable DNA damage[10,68]. Remarkably, MMR components appear to directly interact with the ATR/ATRIP signaling components following DNA damage[69]. The damage signaling process appears far more complex than MMR and the suppression of homeologous recombination since it involves chromatin, chromatin structure and long-range diffusible signaling processes.
1.4. Lynch Syndrome
- The majority of Lynch syndrome or hereditary non-polyposis colorectal cancer (LS/HPNCC) is caused by defects in the human MMR genes[70]. The majority of mutations have been found in the hMSH2 and hMLH1 genes, with approximately 10% of mutations occurring in hMSH6 and hPMS2[70]. MMR defects are thought to drive cancer by elevating mutation rates (Mutator Hypothesis;[71]. However, LS/HNPCC patients are heterozygous for MMR defects while the disease penetrance suggests that affected family members have a greater than 90% chance of developing cancer in their lifetime[70]. This observation suggest that mutation of the remaining normal MMR gene copy may have a “selective advantage”, which should not be the case for simple MMR defects that only increase the rate of spontaneous mutations after the normal copy is mutated. The explanation for this conundrum appears to be the role of MMR in damage signaling. For example, when confronted with excessive DNA damage, cells containing even one copy of the MMR genes will signal apoptosis normally[72]. In contrast, a cell with an MMR defect will tolerate the DNA damage, survive, and then in subsequent cell divisions accumulate mutations from which the numerous genetic alterations that are found in cancer cells can arise[10,68].
1.5. The Development of FRET in MMR Studies
- Until recently, there were no definitive experimental methodologies capable of authenticating the interactions and mechanisms associated with MMR. However, single molecule FRET/Fluorescent Tracking studies (smFRET/FT) have begun to provide substantial quantitative and visual evidence for the Molecular Switch Sliding Clamp MMR model[36,39,73]. These initial results suggest that single molecule analysis has the capability of visualizing and characterizing the entire MMR mechanism. Incorporating FRET into single molecule applications provides extremely accurate nanometer (nm) distance measures between individual protein particles containing either a donor or acceptor fluorophore[74]. The advantage to smFRET/FT includes the ability to image molecules in real-time, the ability to observe intermediate populations, fast acquisition times (usually in the low msec range), and single particle tracking with nm resolution. Latter sections will describe in more detail how the molecular mechanisms associated with the MMR reaction has been advanced using smFRET/FT analysis and what further studies are required.
2. Single Molecule Studies of MMR
2.1. Muts Homologs
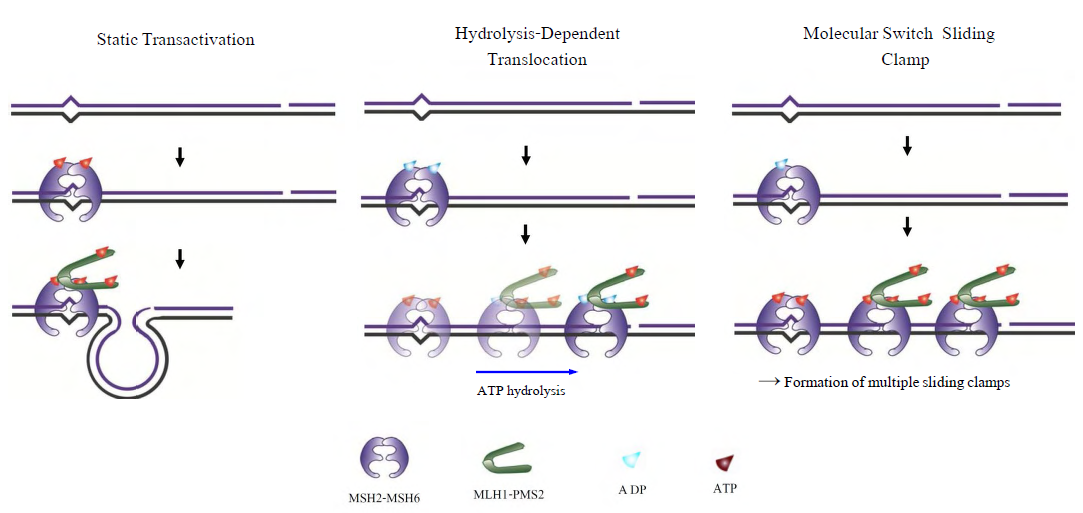
- The functional analysis of the MutS homologs has taken center stage in both bulk and single molecule studies because of its central role in MMR initiation. This section will focus on the historical development of single molecule FRET analysis of MutS homologs with particular focus on results that could not be obtained by any other methodology.Mismatch recognition and its signal transfer – In 2007, the era of MMR single molecule analysis opened when the yeast MutS homologs, ScMsh2-ScMsh6, were shown to diffuse on duplex DNA[75]. These initial observations used a unique smFT system that attached “curtains” of λ DNA to an engineered groove in a flow cell. This allowed the video capture and analysis of hundreds of DNA molecules that were bound across the horizontal groove and were stretched by laminar flow. The ScMsh2 protein within the ScMsh2-ScMsh6 heterodimer contained a His6-tag that could be bound by a anti-His antibody that was labeled with a Quantum Dot (qDot). Movement of the ScMsh2-ScMsh6-antiHis-qDot was tracked on individual λ DNA molecules by prism-based Total Internal Reflection Fluorescent (TIRF) microscopy where the critical angle of an excitation laser is controlled by a prism above the flow cell. An optical filtered charge coupled devise (CCD) camera captured the qDot emission fluorescence through the objective lens of the microscope. These studies visualized diffusion of ScMsh2-ScMsh6 on duplex DNA in a neutral buffer with 50 mM salt. The authors concluded that ScMsh2-ScMsh6 likely diffused on duplex DNA with rotation by comparing the measured diffusion coefficient with theoretical 1D-diffusion with rotation or purely translational 1D-diffusion constants. The result seemed to imply that MutS homologs search for mismatches on duplex DNA by tracking the DNA helix. Remarkably, these studies also observed several ScMsh2-ScMsh6 proteins with very long lifetimes. These observations were used to support a Static Transactivation model. Later studies from the same group[73] as well as others[36,39] have disputed these long-lived species; suggesting they were unusual and/or rare protein events that arose from either the low ionic conditions, the non-equilibrium flow-system or the qDot labeling technique. It is also important to note that the combination of qDot-labeled anti-His antibody required for fluorescent visualization is larger than the ScMsh2-ScMsh6 heterodimeric protein, raising the question of whether the visualization method may have unduly influenced the diffusion analysis from its beginnings. The authors argue against this possibility in supplemental calculations[75]. However, such long-lived MSH species are rarely observed in the absence of flow systems and qDot labeling[36,39]. The ability to observe and trap rare events can be both valuable and misleading with single molecule analysis. Although these authors visualize for the first time how an MSH interacted with duplex DNA, many more questions remained unanswered such as: its behavior in physiological salt (100-150 mM), the lifetime of MSH’s on DNA, the interaction(s) with mismatched DNA, ATP and ADP dependences, among others.To address these specific questions, a unique smFRET system was designed (Fig. 3)[39]. These investigators used Thermus aquaticus MutS (TaqMutS), which containes only one Cys residue (C42) that could be labeled with a single Cy3 fluorophore using maleimide chemistry (Fig. 3A). Unfortunately the protein was completely inactivated by the C42-Cy3 label since it was located in a domain that is used to interrogate mismatched DNA (Fig. 4)[31,32]. To solve this problem, C42 was mutated to an Ala (C42A) that had no effect on TaqMutS function. Then a Thr residue (T469) on the outside surface of the TaqMutS protein was changed to a Cys (T469C) and labeled with Cy3 that had no effect on TaqMutS function (Fig. 3A). Using FRET between Cy3 on TaqMutS and Cy5 on the DNA, the lifetime of TaqMutS on duplex DNA and DNA containing a mismatch was examined in near physiological conditions (100mM salt; Fig. 3). Remarkably, the lifetime of TaqMutS on duplex DNA was 10-fold shorter when the surface-bound DNA contained an open-end compared to when it was blocked with an anti-digoxigenin antibody (0.31s compared to 3.7s; Table 3; Fig. 3B and 3C; TaqMutS lifetime τduplex•off for unblocked DNA may be calculated as the inverse of the kduplex•off = 3.2 s-1). This result strongly suggested that the MutS protein formed an incipient clamp on duplex DNA that was retained when both ends are blocked (biotin-streptavidin on the surface and dig-antidig). While biochemical and structural analysis suggested that MSH proteins formed a clamp when bound with ATP[38] and when bound to a mismatch (Fig. 4)[31,32], this was the first direct evidence that MSH proteins formed a clamp while searching for a mismatch on duplex DNA. It was the short time resolution provided by this smFRET system (30ms) that allowed the lifetime measures of TaqMutS on blocked and unblocked duplex DNA.
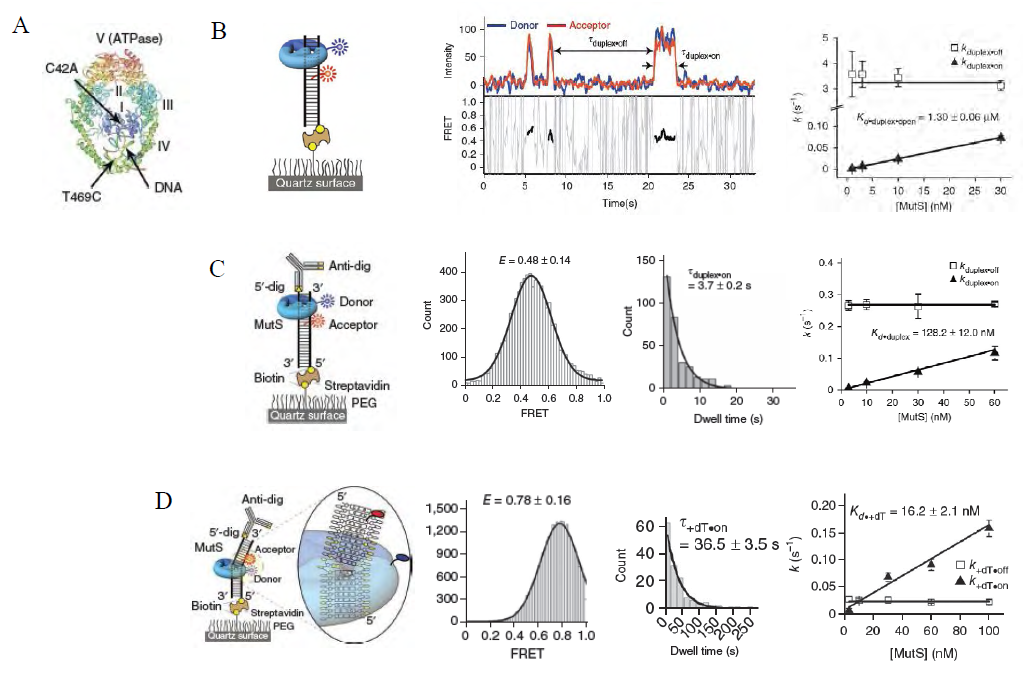 | Figure 3. smFRET Analysis of TaqMutS Association with Duplex DNA. A. Structure of TaqMutS bound to a mismatch showing peptide domains I–V (PDB 1EWQ)42. Donor Cy3 was conjugated to Cys469 of TaqMutS(C42A,T469C). B. (left) Schematic representation of smFRET assay with the duplex DNA containing an open end. Cy5-labeled duplex DNA molecules (74 bp) were immobilized on a quartz surface via a biotin-streptavidin linker. (center) representative kinetic scan showing TaqMutS binding lifetimeduplex•on) and dissociation lifetime (duplex•off). (right) Determination of Kd•duplex•open from kduplex•off (1/duplex•off) that was independent of TaqMutS concentration and the slope of kduplex•on (1/duplex•on) versus TaqMutS protein concentration. C. (left) Schematic representation of smFRET assay with the duplex DNA containing blocked ends. The “free” end is blocked with anti-digoxigenin (anti-dig) and while the remaining end is bound to the surface via a biotin-streptavidin linkage. (center) The FRET efficiency and distributions of binding lifetime for 10 nM MutS in 100 mM KCl. A single exponential with mean ± s.e.m. fit the distribution. (right) The Kd•duplex•blocked determined as in B(right) above. D. (left) Schematic representation of smFRET assay with the mismatched DNA containing blocked ends. (center) The FRET efficiency and distributions of binding lifetime for 10 nM MutS in 100 mM KCl. A single exponential with mean ± s.e.m. fit the distribution. (right) The Kd•mismatch determined as in B(right) above. (taken from Jeong et al., 2011) |
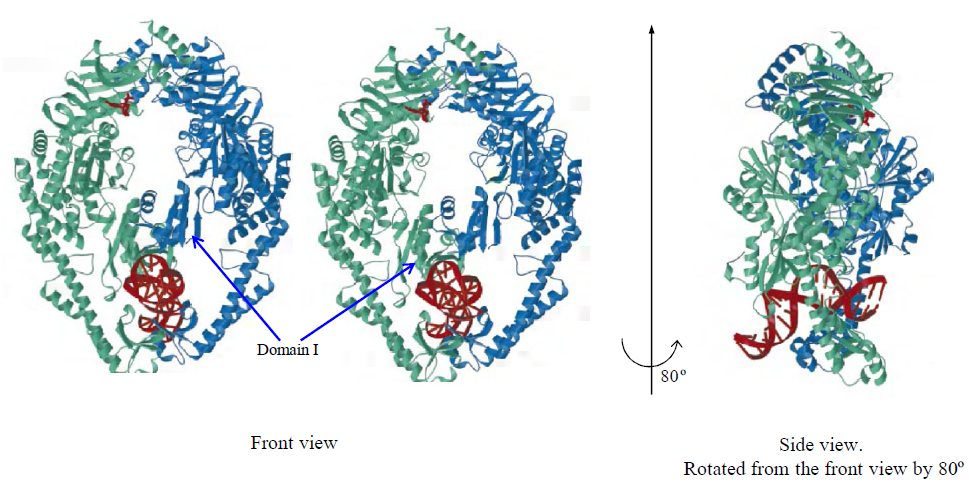 | Figure 4. The structure of MutS binding to mismatched DNA. DNA and ADP are colored red, the mismatch-binding monomer light green, and the second monomer blue A. Front view. Mismatch interrogation Domain I is identified with arrows. B. Side view. Rotated from (A) by 80o |
|
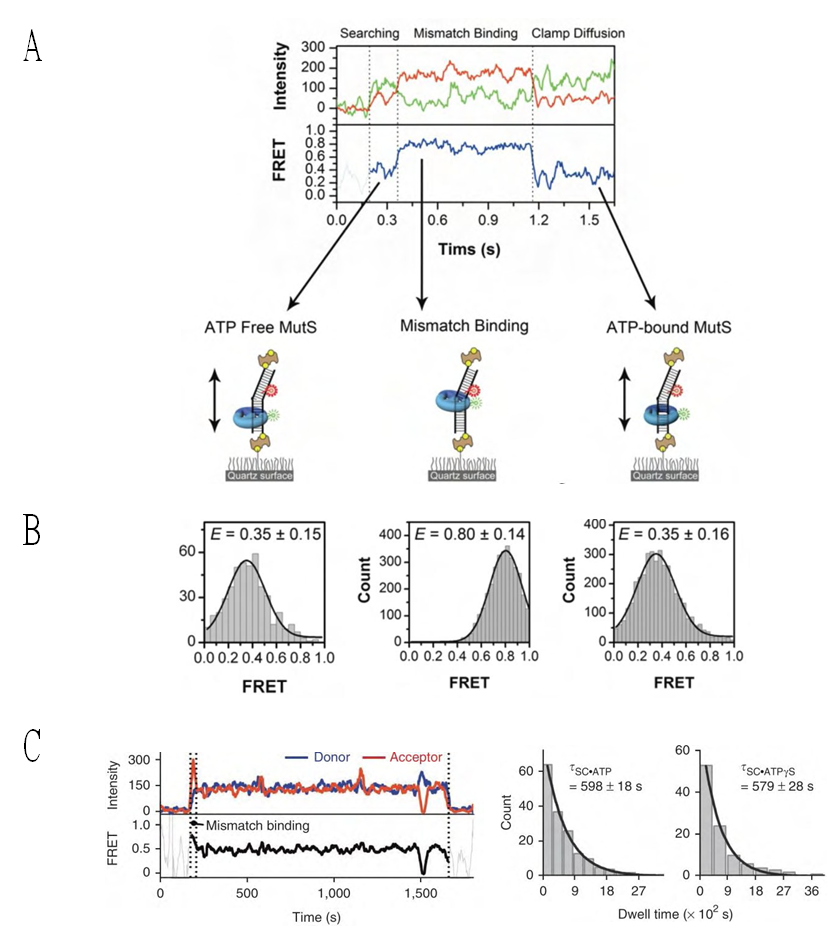 | Figure 5. Kinetic Analysis of TaqMutS Association with Mismatched DNA in the Presence of ATP. A. Representation scan of TaqMutS association with 100 bp DNA containing a +dT mismatch during mismatch searching (ATP free MutS), binding (Mismatch Binding), and ATP processing (ATP-bound MutS). B. Histogram of FRET efficiency during the MutS search (0.35 ± 0.15), binding (0.80 ± 0.14), and ATP–induced clamp diffusion (0.35 ± 0.15) on the 100 bp DNA containing a +dT mismatch, respectively. The error bars are obtained from the s.d. C. (left) Representative time-lapse trace of ATP-bound Taq MutS on the +dT mismatched DNA substrate. (right) Dwell time of the sliding clamp FRET state of MutS in the presence of ATP and ATPγS determined from a single exponential of a population histogram of +dT molecules. (taken from Jeong et al., 2011) |
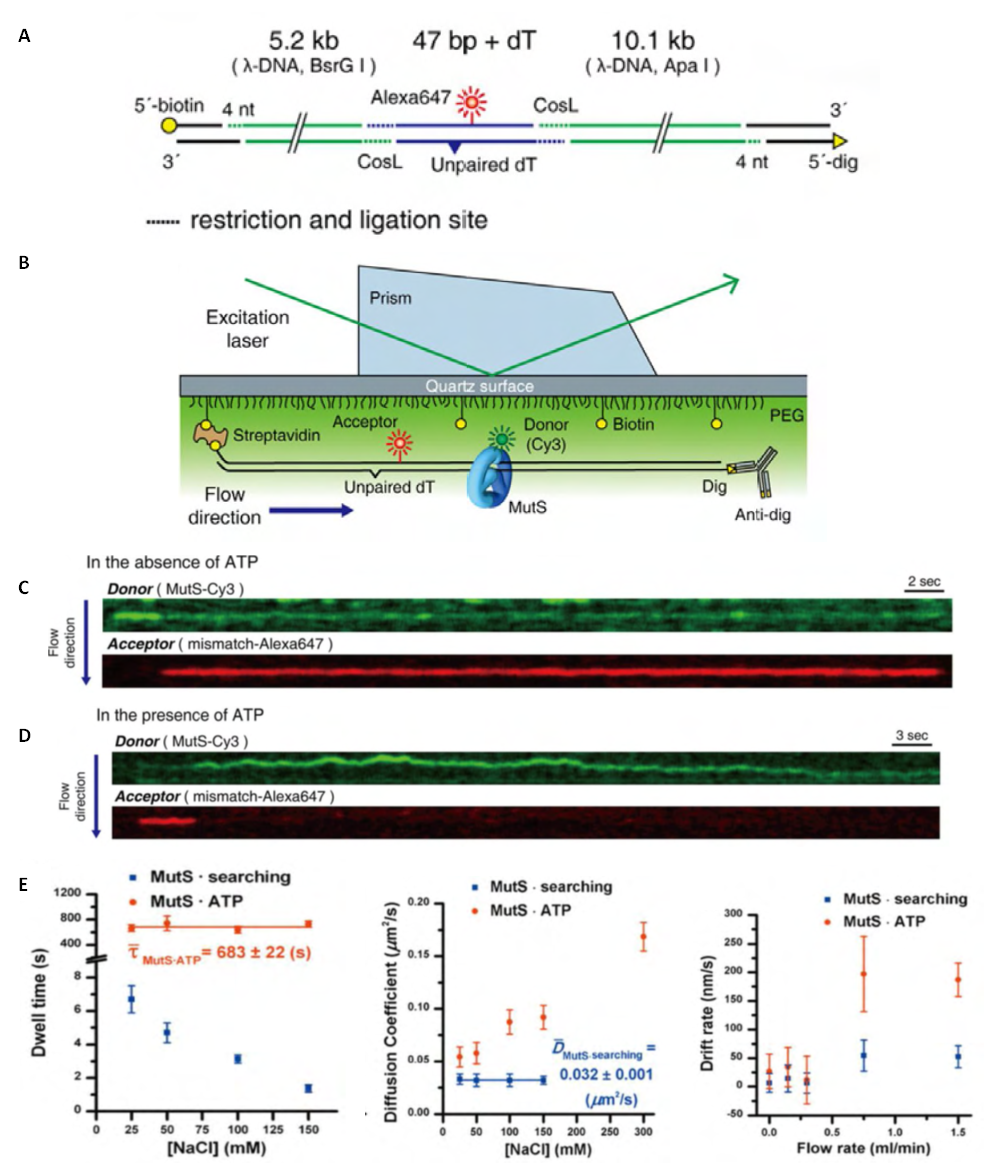 | Figure 6. Single-Molecule Tracking of MutS on DNA. A. Illustration of the 15.3 kb DNA used for smFlow-FRET. B. A schematic representation of smFlow-FRET using prism-type total internal reflection fluorescence (TIRF) microscopy. C. A representative kymograph that shows searching MutS (strong green signal), followed by mismatch binding (reduced green signal; increased red FRET) in the absence of ATP. D. A representative kymograph of Cy3-MutS mismatch interaction(s) in the presence of ATP (200 mM). FRET emission by Alexa647 (red) indicates mismatch binding, followed by the formation of an ATP-bound MutS sliding clamp. E. The Distinct Diffusion Mechanism of Searching MutS and ATP-Bound MutS. (left) The dwell times of the Cy3-MutS on a 100 bp duplex DNA (blue) and ATP-bound MutS on a 100 bp DNA containing an unpaired dT mismatch (red; τ,τ.MutS•ATP = 683 ± 22 s, mean ± s.e.) as a function of ionic strength. For the ATP-bound Cy3-MutS time-lapse smFRET was exploited (see Jeong et al., 2011). (center) The diffusion coefficients of searching Cy3-MutS (blue; ,τMutS•searching = 0.032 ± 0.001 μm2s-1, mean ± s.e.) and ATP-bound Cy3-MutS (red) on the 15.3 kb DNA containing a mismatch at various salt concentrations (25, 50, 100, and 150 mM NaCl). (right) The drift rate of protein trajectories versus flow rate. All error bars indicate s.e. (taken from Cho et al., 2012) |
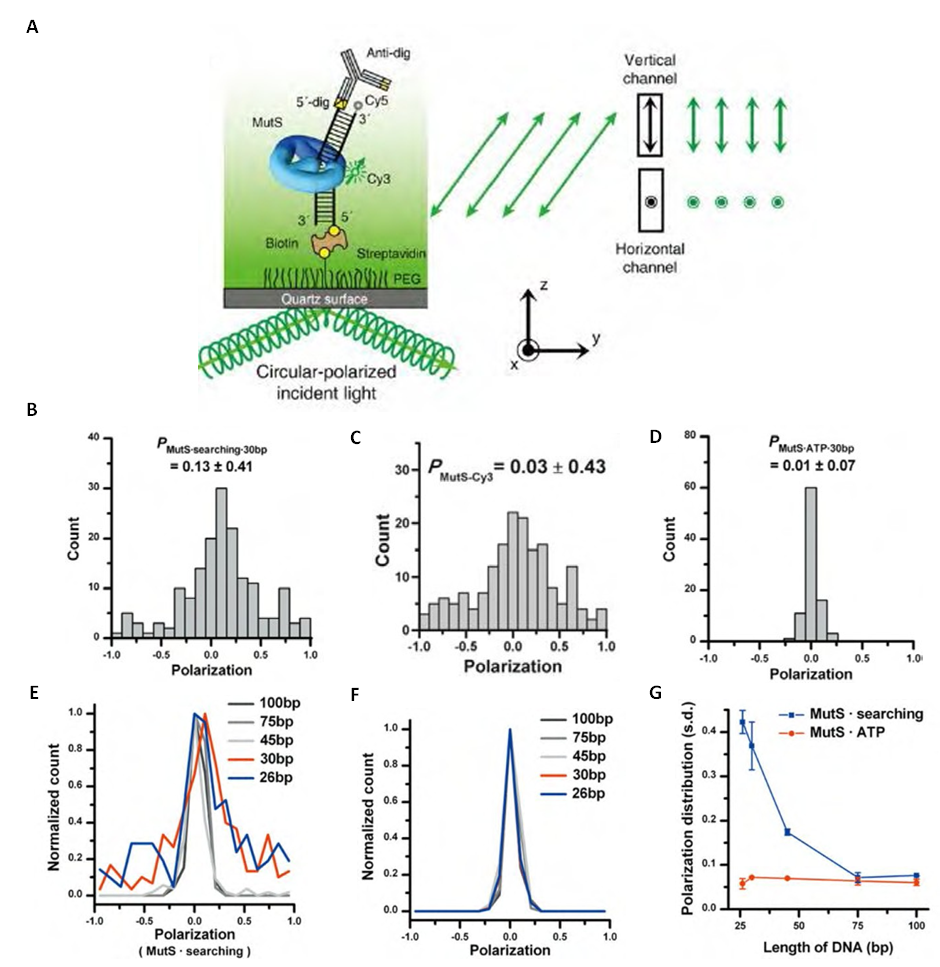 | Figure 7. The Rotational Diffusion of MutS along DNA Using smPolarization-TIRF. A. A schematic representation of the smPolarization-TIRF system. A circularly polarized beam, used to excite Cy3-MutS, was colocalized to the DNA via Cy5 emission. The emission polarization directions are defined as a ‘‘horizontal’’ polarization (IH) in the plane parallel to the microscope stage and a ‘‘vertical’’ polarization (IV) in the plane perpendicular to the microscope stage. B. The steady–state polarization of Cy3-MutS nonspecifically immobilized on the surface, which results in the random distribution of the MutS on the surface. The resulting Cy3-MutS polarization is broadly distributed from –1.0 to 1.0. These results indicate that a rigid linkage to the MutS protein suppresses the rotational freedom of the Cy3. C. The distribution of polarization of Cy3-MutS restricted by DNA length. (PMutS•searching•30bp = 0.13 ± 0.41, mean ± s.d.; n = 170). D. The distribution of polarization of Cy3-MutS unrestricted by DNA length. (PMutS•mismatch•ATP•30bp =0.01 ± 0.07, mean ± s.d.; n = 91). E. The polarization distributions at various lengths (26, 30, 45, 75, and 100 bp) of duplex DNA. F. The polarization distributions at various lengths of duplex DNA containing an unpaired dT mismatch. G. The polarization distribution of searching Cy3-MutS and ATP-bound Cy3-MutS at different lengths. Error bars indicate s.e. (taken from Cho et al., 2012) |
2.2. Mutl Homolog Studies
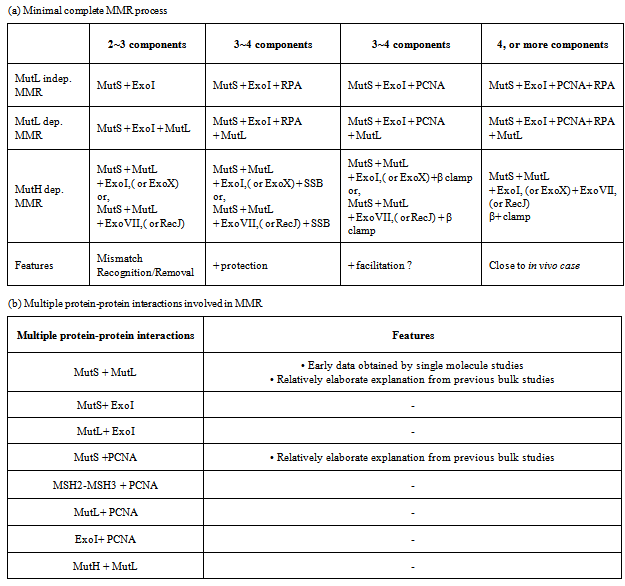
- The function of MLH/PMS proteins in MMR has been enigmatic. Both cellular and biochemical reconstitution studies clearly suggest a central role in licensing the strand scission (E.coli) and/or regulating the strand excision process. MLH/PMS proteins contain a GHKL (DNA Gyrase, Hsp90, Histidine Kinase and MutL) ATP binding and hydrolysis motif[79,80]. When bound with ATP, the N-terminal domains of the MLH/PMS homodimer or heterodimer have been suggested to associate (dimerize) with one another[79]. The C-terminal domains of all MLH/PMS proteins have been shown to form stable complexes with their homodimeric or heterodimeric partner[81,82]. The combination of these two observations suggest a homo- or hetero-dimeric protein that has a stable hinge at the C-terminus, a potentially ATP-dependent clasp at the N-terminus, and a large flexible linker between these two domains. Finally, bulk biochemical studies have suggested the MLH/PMS proteins posses an ATP-dependent ssDNA binding activity of unknown function[83-86]. Single molecule analysis of MLH/PMS proteins – In 2010 two papers appeared in the literature that examined activities of single MLH/PMS proteins on DNA[87,88]. Gorman et al., used the DNA curtain smFT system described above, that were modified to contain nanofabricated anchors 15 nm above a passivated surface, to examine the dynamics of ScMlh1-ScPms1[87]. This extraordinary double anchoring method allowed the observation of protein diffusion along the DNA in the absence of hydrodynamic force; a potentially confounding issue in the DNA curtain system (see above). The association of ScMlh1-ScPms1 with duplex DNA was visualized. Remarkably, the diffusion properties appeared consistent with a ring-like architecture that was independent of adenosine nucleotide binding. Moreover, individual ScMlh1-ScPms1 molecules appeared to bypass one another while traveling along the DNA. Because the diffusion coefficient increased with ionic strength, the authors concluded that movement by ScMlh1-ScPms1 occurred by a hopping or stepping mechanism. A polynucleosome array was reconstituted on the DNA curtains where the individual histone octamers that make of the core of reconstituted nucleosomes could be visualized with a qDot-labeled anti-FLAG antibody bound to FLAG-tagged histone H2A. Diffusing ScMlh1-ScPms1 was observed to easily transit nucleosomes suggesting very large hops/steps. The authors then introduced a TEV protease site into the flexible linker region between the C-terminal clasp and the N-terminal GHKL ATPase domains and found that it significantly disrupted the ability of ScMlh1-ScPms1 to associate with the duplex DNA.Park et al., used an smFRET system to examine ssDNA binding by E.coli MutL (EcMutL)[88]. The system employed a short 15bp duplex DNA containing a 33nt oligo-dT tail. A Cy3-donor fluorophore was placed at the end of the ssDNA tail and a Cy5-acceptor fluorophore was placed at the dsDNA-ssDNA junction. In the absence of protein binding the ssDNA would randomly coil in solution placing the Cy3-Cy5 in relatively close proximity and increasing FRET. When bound by an ssDNA binding protein the oligo-dT33 will be stretched and the distance between the Cy3-Cy5 will increase, resulting in decreased FRET. Under conditions where bulk ssDNA binding was observed (25 mM salt), the addition of MutL was shown to reduce FRET[88]. FRET reduction was increased ~3-fold in the presence of ATP and mutation of a critical ssDNA binding residue[EcMutL(R266E)] resulted in FRET that was nearly equivalent to the absence of protein. To confirm the FRET studies a single molecule flow-stretching (smFS) analysis was developed where a 5.3 Kb ssDNA was attached to the surface at one end and a 2.8μm polystyrene bead attached to the other end. At a fixed hydrodynamic force that was controlled by laminar flow rate, the bead position was fixed and dependent on the retraction forces associated with the random coil of ssDNA. Binding EcMutL uncoils the ssDNA extending the position of the bead in the laminar flow. These studies confirmed EcMutL ssDNA binding, but demonstrated that the binding/extension activity decreased to zero at 120 mM salt.Combining the Gorman et al.,[73] and Park et al.,[88] single molecule studies has begun to illuminate MLH/PMS function(s). One seeming contradiction is that Park et al.,[88] observed essentially no ssDNA binding/extension at 120 mM salt, while Gorman et al., observed relatively stable dsDNA binding and step/hop diffusion in 150 mM salt. These observations might be reconciled if one hypothesizes that both DNA binding domains of the homo-/hetero-dimer located in the flexible linker must associate with the DNA for binding/extension, while only one is required for step/hop diffusion. In such a case, increasing salt would affect the lifetime and dynamics of each binding domain independently ultimately increasing the probability that one will dissociate in the time frame of the smFS analysis. At physiological salt, the MLH/PMS would appear to form some type of very large and flexible clamp that may step/hop using alternate DNA binding domains but is incapable of stably binding/stretching the DNA. Interestingly, later smFT DNA curtain studies appear to suggest that step/hop-diffusing ScMlh1-ScPms1 strongly associates with ATP-bound ScMsh2-ScMsh6 sliding clamps (i.e. it does not step/hop over the ScMsh2-ScMsh6)[73]. These observations are clearly intriguing and require additional single molecule and/or FRET analysis.Although the Gorman et al.,[73] and Park et al.,[88] single molecule studies have opened the door for investigating the cryptic behaviors of MLH/PMS proteins, several questions remain: 1.) what kind of specific interactions occur between the sliding complex of MSH and MLH/PMS and downstream partners to elicit DNA excision, 2.) when and how is the MLH/PMS endonuclease activated, 3.) what is the specific role of multiple MSH-MLH/PMS sliding clamp complexes, 4.) What happens to the MLH/PMS structure when it associates with the ATP-bound MSH sliding clamp, among others. In addition to these MMR questions, there are also basic biophysical questions. The Gorman et al.,[73] studies suggest that the sliding complex containing ScMsh2-ScMsh6/ScMlh1-ScPms1 display a similar dwelling time and a faster diffusion coefficient than ScMSh2-ScMsh6 alone. The similar dwell time might be explained if there is occlusion of the ScMlh1-ScPms2 DNA binding site(s) by the ScMsh2-ScMsh6/ScMlh1-ScPms1 complex. However, a faster diffusion coefficient is opposite to what one would expect for the diffusion of a larger complex with a larger surface area exposed to the viscous solution. Clearly, quantitative smFRET/FT analysis will help to resolve these issues.
3. Technical Development Required for FRET Analysis of MMR
- The ultimate goal of the MMR single molecule FRET studies is the visualization of the complete reaction in both the prokaryotic and eukaryotic systems. Part and parcel to this goal is the analysis of individual protein-protein interactions that occur during MMR. The latter studies appear more technically feasible with present-day technologies since many of these are simple two-component interactions. However, if one is to use smFRET/FT as a method of visualization, a much less obtrusive method for fluorophore labeling will be required than the use of qDot-labeled antibodies. This issue underlines several technical issues that need to be addressed in the development of MMR single molecule studies that include: 1.) Fluorophore labeling of proteins, 2.) Spatial resolution as it applies to the co-localization of protein-protein and protein-DNA interactions, 3.) Rectifying different results from different laboratories, and 4.) Constructing appropriate DNA substrates and visualizing them in an appropriate single molecule apparatus.
3.1. Fluorophore Labeling of Proteins
- The first technical issue is fluorophore labeling of proteins for smFRET/FT analysis. A number of methods have been developed for protein labeling by researchers working in chemistry, biochemistry and biophysics. These methods can be broadly broken down into two major groups: 1.) introduction of specific amino acid modifications amenable to chemical labeling, and 2.) addition of peptide fusion tags. Amino acid modifications include cysteine introduction/deletion, amine group modifications, and non-natural amino acids incorporation. The addition of peptide fusion tags include fluorescent proteins (GFP, RFP, etc.,), Halo(haloalkane dehalogenase)-Tag, SNAP/CLIP (O6-alkylguanine-DNA alkyltransferase)-Tag, Avi(biotin ligase recognition peptide)-Tag, Sfp phosphopantetheinyl transferase(CoA)-Tag, and FGE(formylglycine generating enzyme recognition peptide)-Tag. Each of these labeling technologies have both advantages and disadvantages.Amino acid modification - One of the most common labeling methods is the conjugation of a thiol-reactive group (such as maleimide-fluorophore) to a solvent-accessible cysteine in the protein. For the smFRET/FT studies described above, TaqMutS was labeled by this method[36,39,78,89]. However, if the protein contains multiple cysteine residues, then site-directed mutagenesis must be performed to replace them. For the vast majority of proteins cysteine labeling is not a viable option either due to excessive cysteine residues or the lack of sufficient structural information required to generate ‘Cys light’ substitution mutations.Amine groups may also be exploited for their ability to react with some amine-reactive reagents (such as succinimidyl esters). Unfortunately, this method often results in multiple labeling events per protein because of the high frequency of lysine and arginine residues. Specific modification of the N-terminal α-amine group with succinimidyl ester conjugates because of their low pKa has been reported[90,91]. However, a recent publication pointed out that the specificity of this method might be less than 40%[92].The incorporation of non-natural amino acids is another method to label proteins. Modified aminoacyl tRNA synthetases have been developed to incorporate non-natural amino acids (some containing a fluorophore) into a growing peptide chain via specific codon recognition. These codons only exist in the proteins of interest so that the labeling is specific. This method has been used for protein labeling studies in vivo[93]. However, the incorporation efficiency of the non-natural amino acid appears to be quite low in vivo, such that a cell-free protein synthesis system is often utilized[94]. In spite of the fact that single molecule analysis requires very little fluorophore-labeled protein compared to bulk studies, it appears that the non-natural amino acid incorporation methodology rarely produces sufficient quantities of protein for an accurate determination.Fusion Protein/Peptides – The fusion of a fluorescent protein has been a staple of protein visualization for over two decades[95]. Since the use of green fluorescent protein (GFP) as a bio-marker, other fluorescent mutants (such as YFP, CFP) have been constructed.[96]. These multiple fluorescent protein derivatives display different excitation/emission spectra and relatively strong photostability, which ultimately makes them useful in FRET and single molecule experiments. There are numerous examples of single molecule imaging studies using GFP-protein fusions in vivo (see for example:[97,98]. However, GFP and its derivatives are large peptides (~27kD) that may interfere with the function and/or interaction(s) of the protein under study. In addition, GFP derivatives may form oligomers that could introduce serious artifacts[96]. The Halo-Tag method employs the haloalkane dehalogenase protein (33kD) where a catalytic triad mutation produces an irreversible covalent link between a fluorophore-modified ligand and the protein[99,100]. Commercially available Halo-Tag ligands labeled with coumarin and fluorescein are presently available. The large size of the haloalkane dehalogenase protein fusion may have similar deficiencies to the GFP derivatives described above.The SNAP-tag (20kD) and CLIP-tag (21KD) are labeling systems based on human O6-alkylguanine-DNA alkyltransferase (hAGT). The hAGT catalyzes auto-modification with an alkyl group from its natural substrate O6-alkylguanine or a benzylguanine[101]. There are several commercially available substates which fluorophores, biotin or beads conjugated to benzylguanine (SNAP-tag) or benzylcytosine (CLIP-tag). However, here again the large size of the O6-alkylguanine-DNA alkyltransferase protein fusion may have similar deficiencies to the GFP derivatives described above.Biotinylation has become a useful method to label, purify, or immobilize proteins. In contrast to chemical biotinylation methods, enzymatic biotinylation allows biotin to be linked to exactly one residue present in the protein. The fusion of a fifteen amino acid peptide (Avi-Tag) to the protein of interest provides a specific site for enzymatic biotinylation. The tagged protein may then be incubated with purified biotin ligase (BirA) in the presence of biotin and ATP in vitro, or co-expressed with the BirA gene in vivo to produce a protein with a single specific biotin residue[102,103]. A biotinylated-protein may then be labeled with a fluorescent streptavidin (53kD) or a fluorescent qDot linked to streptavidin (>53kD). Biotinylated proteins are extremely rare in nature, making the chances for cross-reactivity very low. The shortcoming of this method is that the size of the conjugated complex may be too large (>15nm) for most proteins (see above). However, biotinylation is clearly a viable method for the immobilization of proteins on a single molecule surface.A small tetracysteine tag has also been used for protein labeling[104,105]. The tetracysteine binding-motif contains six amino acids (CCXXCC), which in most cases is unlikely to disrupt native protein function. Invitrogen currently markets fluorescent molecules that may be specifically linked to the tetracysteine tag. However, non-specific binding of the fluorescent molecules to other cysteine rich motifs makes this approach unsuitable for many single molecule applications[106,107].Sfp phosphopantetheinyl transferase recognizes the ybbR tag (eleven amino acids) and will transfer a 4’-phos-phopantetheinyl group from CoA to a serine residue in the tag[108,109]. Proteins with ybbR tags can be specifically labeled using fluorescent CoA. SFP Synthase and fluorescent CoA (CoA-488, CoA-547 and CoA647) are now available from New England Biolabs, and several single molecule studies have used this method[110-113]. One drawback of this method is the labeling efficiency. Unlike the FGE method in which tag modification and tag labeling are two distinct steps, this labeling protocol is done in one intricate step. The requirement that the modification be done in vitro and at low temperature makes for difficult enzymology. Moreover, the reported labeling to date is 17% after 24h[110].Formylglycine generating enzyme (FGE) catalyzes the formation of a formylglycine residue from a central cysteine in a highly conserved six amino acid motif (LCTPSR; [92,114,115]. The formylglycine post-translational modification may be introduced in vivo by co-expression of FGE or in vitro using purified FGE protein[92,114,115]. Aminooxy- or hydrazide-functionalized probes may be conjugated to the aldehyde group of formylglycine. The reaction is specific as there are very few aldehyde groups in naturally occurring amino acids. It has been reported that 100% protein labeling can be obtained using this method, and is suitable for single molecule experiments[92]. The shortcoming of this method is that a large amount of dye must be used[92] and the spontaneous reversibility of the hydrozone linkage[116].These labeling methods present many opportunities for measuring proteins in smFRET/FT systems. Specific labeling is particularly important for smFRET, in which two fluorophores at defined distance are required. Although it is possible to purify and dual-label proteins in vitro, many problems still remain with the labeling methods themselves. A new method that has higher efficiency, greater specificity, and does not interfere with normal protein function, would greatly expand the application of single molecule experiments in biological events.
3.2. Spatial Resolution and Co-localization of Interactions
- Monitoring multiple proteins simultaneously and eventually the entire MMR process presents significant resolution challenges. Because the spatial resolution of smFRET/FT is diffraction limited (~200-300nm) and the typical footprint of an MSH is about 10-20nm, it is possible that there may be over 10 ATP-bound MSH sliding clamps in one florescent spot recorded by the CCD camera. Gorman et al.,[75] found that with a fixed Qdot the standard deviation was about +/- 16nm with a 2D Gaussian peak analysis of a fluorescent spot in the DNA curtain smFT[75]. However, as a result of the fluctuating nature of long DNAs in smFT, proteins in thermal motion and the properties of the fluorophore, it is inferred that the spatial resolution is likely to easily exceed 20nm; even from the 2D Gaussian peak analysis where there may be several MSH molecules in one spot. These observations are likely to cloud any conclusions whether a protein-protein interaction on an smFT DNA is coincident or specific. Several schemes have been devised to overcome this obstacle. For example, bleaching fluorophores to count the number of molecules, measuring speed, observing collision, and designing multiple FRET pairs.Super resolution microscopy that resolves single molecules below the diffraction limit has begun to come of age using RapidSTORM (Neubeck& Van Gool algorithm) [117], QuickPALM (classical Högbom ‘CLEAN’ algorithm) [118], LivePALM (fluoroBancroft algorithm) [119], radial symmetry centers[120] or Maximum Likelihood Estimation (MLE) algorithms[121] to localize particles. Most supper resolution studies have required fixed samples since the number of photons is usually quite low and any molecular movement (diffusion) would influence the acquisition algorithms ultimately decreasing the special resolution. Work on feed back mechanisms that rely on parallel localization processing continues to show promise for super resolution of particle locaization in real time[122].
3.3. Variation in the Quantitative Analysis between Multiple Laboratories
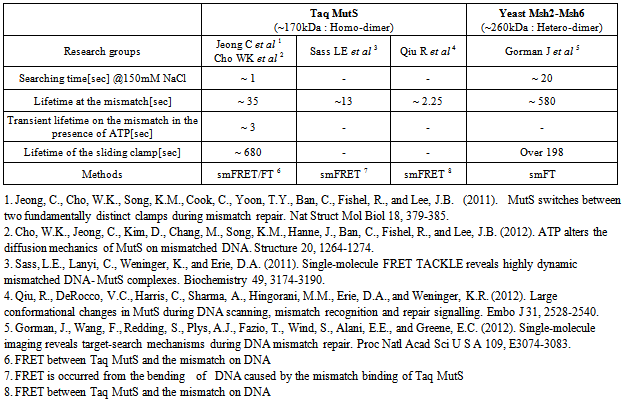
- The differences in quantitative analysis between laboratories are most clearly demonstrated in Table 3. It might be argued that differences between TaqMutS and ScMsh2-ScMsh6 could be explained by the size differences and the nature of the complex (homodimer vs. heterodimer). However, the wide range of measures, even between different TaqMutS studies is disturbing. There are some factors that might have affected results that include the use of a relatively strong flow and the use of differing ionic conditions. Careful comparison of experimental procedures and designs might explain some if not all of the disagreements. One issue that would clearly help to mollify differences would be to insist that single molecule studies be performed under physiologically relevant ionic conditions.
3.4. Construction of Appropriate DNA Substrates and Visualization
- The cellular process of MMR occurs on DNA with mismatches, nicks, gaps and/or methylated sites for E.coli as well as in the presence of fully or partially reconstituted chromatin. Due to the spatial resolution problem a long DNA is required to observe a complete process. However, it is not a simple task to construct a DNA that is over 10kb and containing a single mismatch. Moreover, before and after the actual single molecule experiment, it is necessary to confirm the status of a long DNA since they are relatively fragile in a flow system. Sytox staining has been typically used to confirm nature of the DNA on a single molecule surface[123]. However, when examining DNA repair proteins that recognize lesions in the DNA, it is important to determine that residual visualizing reagent does not interfere with the observations. Despite these obstacles smFRET/FT studies of MMR components have been successful. There is no doubt that with some small improvements, the smFRET/FT methods will become more promising schemes in studying MMR.
4. Concluding Remarks
- FRET studies have played an important role in detailing the interactions and kinetics of MMR proteins on mismatched DNA. Careful design of FRET donor and acceptor locations is essential to insure accuracy as well as a lack of influence on biological functions. We have provided several examples where the lack of careful attention to biophysical details has likely led to dramatic overinterpretation of the data and models with almost no biological relevance. Clearly the development of labeling technologies and new quantitative analysis will lead to further understanding of the biophysics of MMR. In addition, the development of real-time cellular single molecule analysis where FRET plays an integral role in determining interactions and positioning is on the horizon.
ACKNOWLEDGEMENTS
- The authors would like to thank members of the Lee and Fishel laboratories for helpful discussions. This work was supported by the National Research Foundation (NRF) of Korea and the Ministry of Education, Science, and Technology (MEST; 2011-0013901; J-BL) and NIH grant CA67007 (RF).