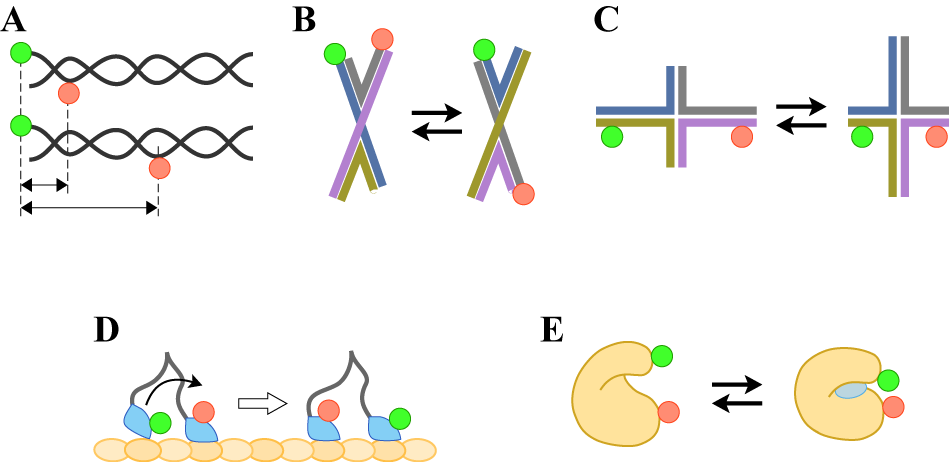| [1] | Selvin, P. R., 2000, The renaissance of fluorescence resonance energy transfer., Nat. Struct. Biol., 7(9), 730–734. |
| [2] | Förster, V. T., 1946, Energiewanderung und Fluoreszenz., Die Naturwissenschaften, 33(6), 166–175. |
| [3] | Stryer, L., and Haugland, R. P., 1967, Energy transfer: A spectroscopic ruler., Proc. Natl. Acad. Sci. USA, 58(2), 719–726. |
| [4] | Haas, E., Katchalski-Katzir, E., Steinberg, I. Z., 1978, Effect of the orientation of donor and acceptor on the probability of energy transfer involving electronic transitions of mixed polarization., Biochemistry, 17(23),5064–5070. |
| [5] | Trautman, J. K., Macklin, J. J., Brus, L. E., Betzig, E., 1994, Near-field spectroscopy of single molecules at room temperature., Nature, 369, 40–42. |
| [6] | Xie, X. S., and Dunn, R. C., 1994, Probing single molecule dynamics., Science, 265, 361–364. |
| [7] | Ambrose, W. P., Goodwin, P. M., Martin, J. C., Keller, R. A., 1994, Single molecule detection and photochemistry on a surface using near-field optical excitation., Phys. Rev. Lett., 72(1), 160–163. |
| [8] | Macklin, J. J., Trautman, J. K., Harris, T. D., Brus, L. E., 1996, Imaging and time-resolved spectroscopy of single molecules at an interface., Science, 272, 255–258. |
| [9] | Trautman, J. K., and Macklin, J. J., 1996, Time-resolved spectroscopy of single molecules using near-field and far-field optics., Chem. Phys., 205(1-2), 221–229. |
| [10] | Funatsu, T., Harada, Y., Tokunaga, M., Saito, K., Yanagida, T., 1995, Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution., Nature, 374, 555–559. |
| [11] | Sase, I., Miyata, H., Corrie, J. E. T., Craik, J. S., Kinosita, Jr., K., 1995, Real time imaging of single fluorophores on moving actin with an epifluorescence microscope., Biophys. J., 69(2), 212–218. |
| [12] | Schmidt, T., Schuetz, G. J., Baumgartner, W., Gruber, H. J., Schindler, H., 1995, Characterization of photophysics and mobility of single molecules in a fluid lipid membrane., J. Phys. Chem., 99(49), 17662–17668. |
| [13] | Dickson, R. M., Norris, D. J., Tzeng, Y.-L., Moerner, W. E., 1996, Three-dimensional imaging of single molecules solvated in pores of poly(acrylamide) gels., Science, 274, 966–968. |
| [14] | Nie, S., Chiu, D. T., Zare, R. N., 1994, Probing individual molecules with confocal fluorescence microscopy., Science, 266, 1018–1021. |
| [15] | Keller, R. A., Ambrose, W. P., Goodwin, P. M., Jett, J. H., Martin, J. C., Wu, M., 1996, Single-molecule fluorescence analysis in solution., Appl. Spectrosc., 50(7), 12A–32A. |
| [16] | Ha, T., Enderle, T., Ogletree, D. F., Chemla, D. S., Selvin, P. R., Weiss, S., 1996, Probing the interaction between two single molecules: Fluorescence resonance energy transfer between a single donor and a single acceptor., Proc. Natl. Acad. Sci. USA, 93(3), 6264–6268. |
| [17] | Ha, T., Ting, A. Y., Liang, J., Caldwell, W. B., Deniz, A. A., Chemla, D. S., Schultz, P. G., Weiss, S., 1999, Single-molecule fluorescence spectroscopy of enzyme conformational dynamics and cleavage mechanism., Proc. Natl. Acad. Sci. USA, 96(3), 893–898. |
| [18] | Ha, T., Zhuang, X., Kim, H. D., Orr, J. W., Williamson, J. R., Chu, S., 1999, Ligand-induced conformational changes observed in single RNA molecules., Proc. Natl. Acad. Sci. USA, 96(16), 9077–9082. |
| [19] | Jia, Y., Talaga, D. S., Lau, W. L., Lu, H. S. M., DeGrado, W. F., Hochstrasser, R. M., 1999, Folding dynamics of single GCN-4 peptides by fluorescence resonant energy transfer confocal microscopy., Chem. Phys., 247, 69–83. |
| [20] | Fries, J. R., Brand, L., Eggeling, C., Köllner, M., Seidel, C. A. M., 1998, Quantitative identification of different single molecules by selective time-resolved confocal fluorescence spectroscopy., J. Phys. Chem. A, 102(33), 6601–6613. |
| [21] | Deniz, A. A., Dahan, M., Grunwell, J. R., Ha, T., Faulhaber, A. E., Chemla, D. S., Weiss, S., 1999, Single-pair fluorescence resonance energy transfer on freely diffusing molecules: Observation of Förster distance dependence and subpopulations., Proc. Natl. Acad. Sci. USA, 96(7), 3670–3675. |
| [22] | Dahan, M., Deniz, A. A., Ha, T., Chemla, D. S., Schultz, P. G., Weiss, S., 1999, Ratiometric measurement and identification of single diffusing molecules., Chem. Phys., 247(1), 85–106. |
| [23] | Deniz, A. A., Laurence, T. A., Beligere, G. S., Dahan, M., Martin, A. B., Chemla, D. S., Dawson, P. E., Schultz, P. G., Weiss, S., 2000, Single-molecule protein folding: Diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2., Proc. Natl. Acad. Sci. USA, 97(10), 5179–5184. |
| [24] | Sako, Y., Minoguchi, S., Yanagida, T., 2000, Single- molecule imaging of EGFR signalling on the surface of living cells., Nat. Cell Biol., 2(3), 168–172. |
| [25] | Weiss, S, 1999, Fluorescence spectroscopy of single biomolecules., Science, 283, 1676–1683. |
| [26] | Ha, T, 2001, Single-molecule fluorescence resonance energy transfer., Methods, 25(1), 78–86. |
| [27] | Roy, R., Hohng, S., Ha, T., 2008, A practical guide to single-molecule FRET., Nat. Methods, 5(6), 507–516. |
| [28] | J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd ed., New York, USA: Springer, 2006. |
| [29] | McCann, J. J., Choi, U. B., Zheng, L., Weninger, K., Bowen, M. E., 2010, Optimizing methods to recover absolute FRET efficiency from immobilized single molecules., Biophys. J., 99(3), 961–970. |
| [30] | Okamoto, K. and Sako, Y., 2012, Variational Bayes analysis of a photon-based hidden Markov model for single-molecule FRET trajectories., Biophys. J., 103(6), 1315–1324. |
| [31] | Hanson, J. A. and Yang, H., 2008, Quantitative evaluation of cross correlation between two finite-length time series with applications to single-molecule FRET., J. Phys. Chem. B, 112(44), 13962–13970. |
| [32] | Schuler, B., Lipman, E. A., Eaton, W. A., 2002, Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy., Nature, 419, 743–748. |
| [33] | Gopich, I. V., and Szabo, A., 2003, Single-macromolecule fluorescence resonance energy transfer and free-energy profiles., J. Phys. Chem. B, 107(21), 5058–5063. |
| [34] | Gopich, I. V., and Szabo, A., 2007, Single-molecule FRET with diffusion and conformational dynamics., J. Phys. Chem. B, 111(44), 12925–12932. |
| [35] | Boukobza, E., Sonnenfeld, A., Haran, G., 2001, Immobilization in surface-tethered lipid vesicles as a new tool for single biomolecule spectroscopy., J. Phys. Chem. B, 105(48), 12165–12170. |
| [36] | Okamoto, K., and Terazima, M., 2008, Distribution analysis for single molecule FRET measurement., J. Phys. Chem. B, 112(24), 7308–7314. |
| [37] | Kapanidis, A. N., Lee, N. K., Laurence, T. A., Doose, S., Margeat, E., Weiss, S., 2004, Fluorescence-aided molecule sorting: Analysis of structure and interactions by alternating-laser excitation of single molecules., Proc. Natl. Acad. Sci. USA, 101(24), 8936–8941. |
| [38] | Terentyeva, T. G., Engelkamp, H., Rowan, A. E., Komatsuzaki, T., Hofkens, J., Li, C.-B., Blank, K., 2012, Dynamic disorder in single-enzyme experiments: Facts and artifacts., ACS Nano, 6(1), 346–354. |
| [39] | Liu, Y., Park, J., Dahmen, K. A., Chemla, Y. R., Ha, T., 2010, A comparative study of multivariate and univariate hidden Markov modelings in time-binned single-molecule FRET data analysis., J. Phys. Chem. B, 114(16), 5386–5403. |
| [40] | McKinney, S. A., Joo, C., Ha, T., 2006, Analysis of single-molecule FRET trajectories using hidden Markov modeling., Biophys. J., 91(5), 1941–1951. |
| [41] | Bronson, J. E., Fei, J., Hofman, J. M., Gonzalez Jr., R. L., Wiggins, C. H., 2009, Learning rates and states from biophysical time series: A Bayesian approach to model selection and single-molecule FRET data., Biophys. J., 97(12), 3196–3205. |
| [42] | Xu, C. S., Kim, H., Hayden, C. C., Yang, H., 2008, Joint statistical analysis of multichannel time series from single quantum dot-(Cy5)n constructs., J. Phys. Chem. B, 112(19), 5917–5923. |
| [43] | Ensign, D. L., and Pande, V. S., 2010, Bayesian detection of intensity changes in single molecule and molecular dynamics trajectories., J. Phys. Chem. B, 114(1), 280–292. |
| [44] | Baba, A., and Komatsuzaki, T., 2007, Construction of effective free energy landscape from single-molecule time series., Proc. Natl. Proc. Sci. USA, 104(49), 19297–19302. |
| [45] | Pirchi, M., Ziv, G., Riven, I., Cohen, S. S., Zohar, N., Barak, Y., Haran, G., 2011, Single-molecule fluorescence spectroscopy maps the folding landscape of a large protein., Nat. Commun., 2, 493. |
| [46] | Schuler, B., Lipman, E. A., Steinbach, P. J., Kumke, M., Eaton, W. A., 2005, Polyproline and the “spectroscopic ruler” revisited with single-molecule fluorescence., Proc. Natl. Acad. Sci. USA, 102(8), 2754–2759. |
| [47] | McKinney, S. A., Déclais, A.-C., Lilley, D. M. J., Ha, T., 2003, Structural dynamics of individual Holliday junctions., Nat. Struct. Biol., 10(2), 93–97. |
| [48] | Hohng, S., Joo, C., Ha, T., 2004, Single-molecule three-color FRET., Biophys. J., 87(2), 1328–1337. |
| [49] | Sarkar, S. K., Andoy, N. M., Benítez, J. J., Chen, P. R., Kong, J. S., Chen, C. H., 2007, Engineered Holliday junctions as single-molecule reporters for protein-DNA interactions with application to a MerR-family regulator., J. Am. Chem. Soc., 129(41), 12461–12467. |
| [50] | Karymov, M. A., Chinnaraj, M., Bogdanov, A., Srinivasan, A. R., Zheng, G., Olson, W. K., Lyubchenko, Y. L., 2008, Structure, dynamics, and branch migration of a DNA Holliday junction: A single-molecule fluorescence and modeling study., Biophys. J., 95(9), 4372–4383. |
| [51] | Blanchard, S. C., Gonzalez Jr., R. L., Kim, H. D., Chu, S., Puglisi, J. D., 2004, tRNA selection and kinetic proofreading in translation., Nat. Struct. Mol. Biol., 11(10), 1008–1014. |
| [52] | Blanchard, S. C., Kim, H. D., Gonzalez Jr., R. L., Puglisi, J. D., Chu, S., 2004, tRNA dynamics on the ribosome during translation., Proc. Natl. Acad. Sci. USA, 101(35), 12893–12898. |
| [53] | Rhoades, E., Cohen, M., Schuler, B., Haran, G., 2004, Two-state folding observed in individual protein molecules., J. Am. Chem. Soc., 126(45), 14686–14687. |
| [54] | Merchant, K. A., Best, R. B., Louis, J. M., Gopich, I. V., Eaton, W. A., 2007, Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations., Proc. Natl. Acad. Sci. USA, 104(5), 1528–1533. |
| [55] | Chung, H. S., Louis, J. M., Eaton, W. A., 2009, Experimental determination of upper bound for transition path times in protein folding from single-molecule photon-by-photon trajectories., Proc. Natl. Acad. Sci. USA, 106(29), 11837–11844. |
| [56] | Warshaw, D. M., Kennedy, G. G., Work, S. S., Krementsova, E. B., Beck, S., Trybus, K. M., 2005, Differential labeling of myosin V heads with quantum dots allows direct visualization of hand-over-hand processivity., Biophys. J., 88(5), L30– L32. |
| [57] | Tomishige, M., Stuurman, N., Vale, R. D., 2006, Single-molecule observations of neck linker conformational changes in the kinesin motor protein., Nat. Struct. Mol. Biol., 13(10), 887–894. |
| [58] | Yasuda, R., Masaike, T., Adachi, K., Noji, H., Itoh, H., Kinosita, Jr., K., 2003, The ATP-waiting conformation of rotating F1-ATPase revealed by single-pair fluorescence resonance energy transfer., Proc. Natl. Acad. Sci. USA, 100(16), 9314–9318. |
| [59] | Kozuka, J., Yokota, H., Arai, Y., Ishii, Y., Yanagida, T., 2006, Dynamic polymorphism of single actin molecules in the actin filament., Nat. Chem. Biol., 2(2), 83–86. |
| [60] | Hanson, J. A., Duderstadt, K., Watkins, L. P., Bhattacharyya, S., Brokaw, J., Chu, J.-W., Yang, H., 2007, Illuminating the mechanistic roles of enzyme conformational dynamics., Proc. Natl. Acad. Sci. USA, 104(46), 18055–18060. |
| [61] | He, Y., Li, Y., Mukherjee, S., Wu, Y., Yan, H., Lu, H. P., 2011, Probing single-molecule enzyme active-site conformational state intermittent coherence., J. Am. Chem. Soc., 133(36), 14389–14395. |
| [62] | Berezhna, S. Y., Gill, J. P., Lamichhane, R., Millar, D. P., 2012, Single-molecule Förster resonance energy transfer reveals an innate fidelity checkpoint in DNA polymerase I., J. Am. Chem. Soc., 134(27), 11261–11268. |
| [63] | Müller-Späth, S., Soranno, A., Hirschfeld, V., Hofmann, H., Rüegger, S., Reymond, L., Nettels, D., Schuler, B., 2010, Charge interactions can dominate the dimensions of intrinsically disordered proteins., Proc. Natl. Acad. Soc. USA, 107(33), 14609–14614. |
| [64] | Murakoshi, H., Iino, R., Kobayashi, T., Fujiwara, T., Ohshima, C., Yoshimura, A., Kusumi, A., 2004, Single-molecule imaging analysis of Ras activation in living cells., Proc. Natl. Acad. Sci. USA, 101(19), 7317–7322. |
| [65] | Huppa, J. B., Axmann, M., Mörtelmaier, M. A., Lillemeier, B. F., Newell, E. W., Brameshuber, M., Klein, L. O., Schütz, G. J., Davis, M. M., 2010, TCR–peptide–MHC interactions in situ show accelerated kinetics and increased affinity., Nature, 9, 963–967. |
| [66] | Hibino, K., Shibata, T., Yanagida, T., Sako, Y., 2009, A RasGTP-induced conformational change in C-RAF is essential for accurate molecular recognition., Biophys. J., 97(5), 1277–1287. |
| [67] | Sakon, J. J., and Weninger, K. R., 2010, Detecting the conformation of individual proteins in live cells., Nat. Methods, 7(3), 203–205. |
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML
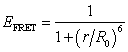
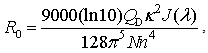
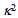 is the orientation factor, which reflects the relative orientation in space of fluorophores and will be mentioned below.
is the orientation factor, which reflects the relative orientation in space of fluorophores and will be mentioned below. 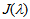 represents the overlap integral, which expresses the degree of spectral overlap between the donor emission and the acceptor absorption. R0 represents the distance, at which the transfer efficiency is 0.5 and is typically 5–10 Å for opt-used dye pairs. The term of 6th power seen in (1) indicates the strong dependence of EFRET on r around R0.EFRET can be calculated as the fraction of the energy transferred to the acceptor in the total energy absorbed by the donor. When fluorescence is detected from a single pair of FRET dyes on two wavelength-separated detector channels, the apparent FRET efficiency Eapp may be approximated with the ratio of detected intensities as
represents the overlap integral, which expresses the degree of spectral overlap between the donor emission and the acceptor absorption. R0 represents the distance, at which the transfer efficiency is 0.5 and is typically 5–10 Å for opt-used dye pairs. The term of 6th power seen in (1) indicates the strong dependence of EFRET on r around R0.EFRET can be calculated as the fraction of the energy transferred to the acceptor in the total energy absorbed by the donor. When fluorescence is detected from a single pair of FRET dyes on two wavelength-separated detector channels, the apparent FRET efficiency Eapp may be approximated with the ratio of detected intensities as 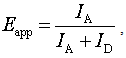
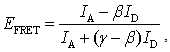
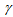 is the coefficient to correct the first factor, the ratio of the detection efficiency between dyes and
is the coefficient to correct the first factor, the ratio of the detection efficiency between dyes and 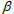 is for compensation of the second factor, the fluorescence leakage[22,29,30].
is for compensation of the second factor, the fluorescence leakage[22,29,30]. 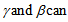 be determined experimentally. If the detected wavelength is restricted by, for example, a band pass filter before the acceptor detector,
be determined experimentally. If the detected wavelength is restricted by, for example, a band pass filter before the acceptor detector, 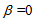 can be assumed at the expense of a portion of photons.
can be assumed at the expense of a portion of photons. 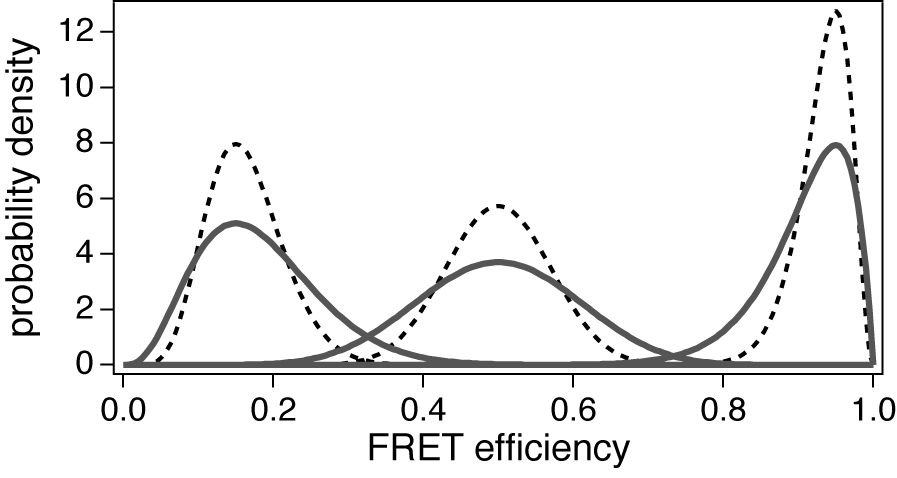
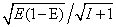 , where E =
, where E = 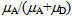 and
and 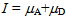 are the mean FRET efficiency and the intensity, respectively, with the average of detected photon counts for the acceptor
are the mean FRET efficiency and the intensity, respectively, with the average of detected photon counts for the acceptor 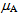 and the donor
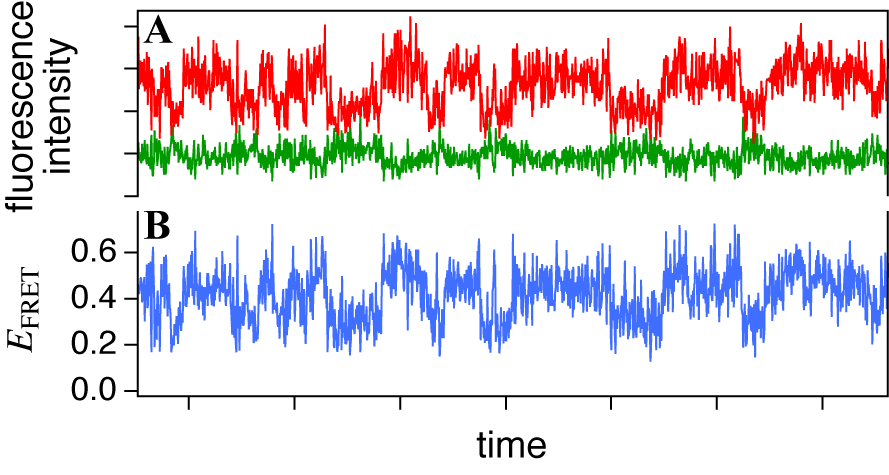
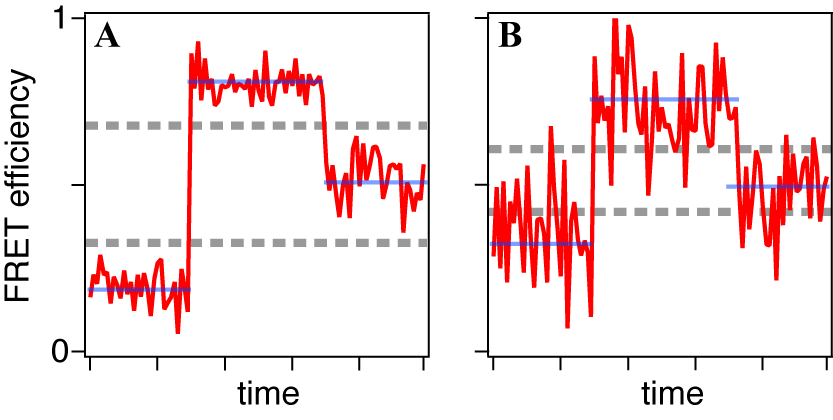
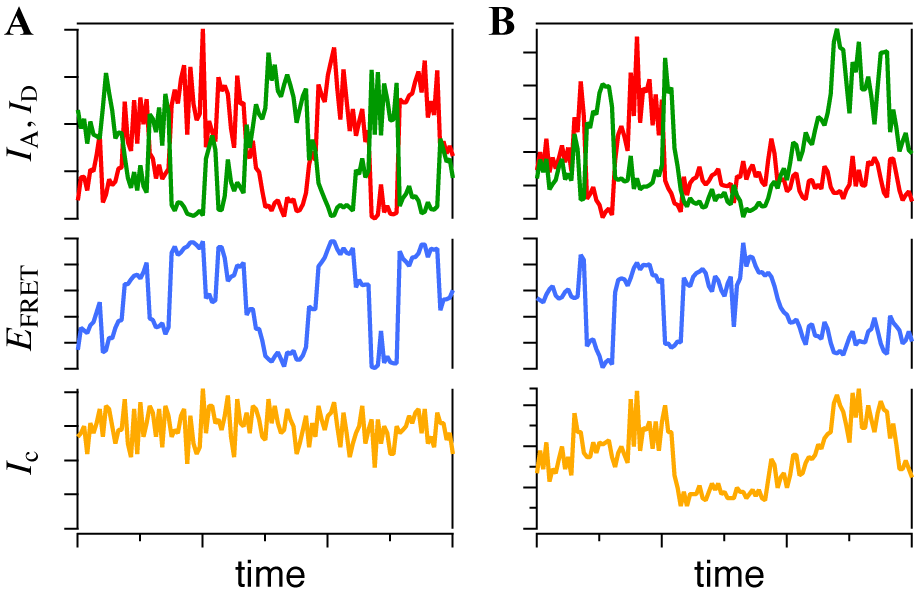
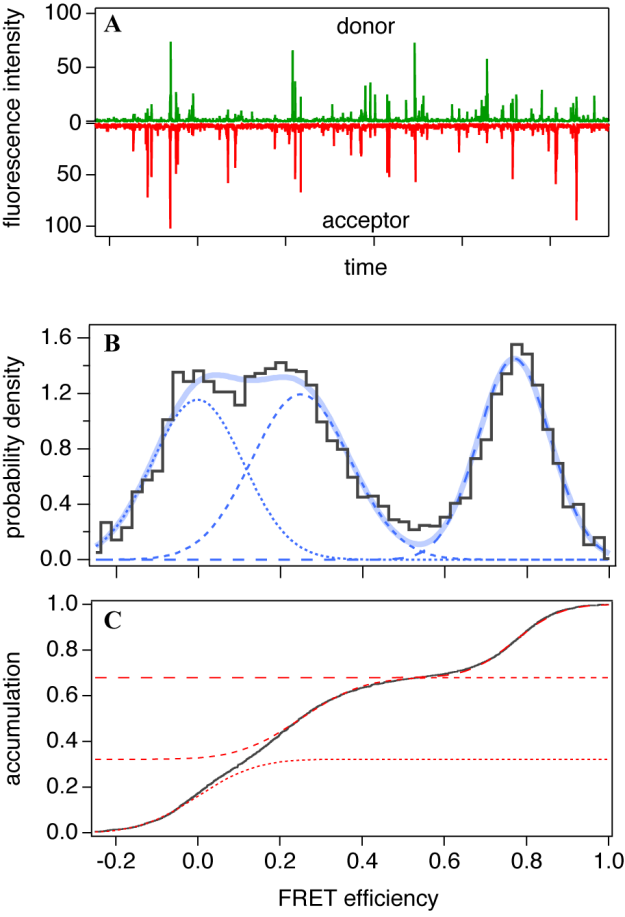
and the donor 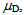 respectively[22]. It suggests that larger I gives a sharper peak and if I is same, the peak becomes sharper near the boundary (E = 0, 1) compared to at the center (E = 0.5). It should be kept in mind that if the compensation of (4) is applied, the Beta distribution describes the distribution before compensation.One of the advantages of smFRET measurement is the ability to trace the time evolution of the FRET changes in realtime like shown in Figures 2. If the FRET efficiency varies with changes in the interdye distance, the acceptor intensity increases while the donor intensity decrease, or vice versa (Figure 2A). However, the fluorescence intensity can be modulated by other reasons than FRET, such as photochemical effects like blinking or photobleach, which are explicitly observed in the single-molecule measurements. When such fluctuations occur, the anti-correlation relationship of the fluorescence intensities is diminished (Figure 2B). In order to distinguish the intensity modulation due to the FRET changes from those instabilities, calculating the cross correlation between two intensity signals was proposed to quantitatively evaluate the anti-correlation relationship[31]. Or more simply, the compensated total intensity Ic can be calculated as[30]
respectively[22]. It suggests that larger I gives a sharper peak and if I is same, the peak becomes sharper near the boundary (E = 0, 1) compared to at the center (E = 0.5). It should be kept in mind that if the compensation of (4) is applied, the Beta distribution describes the distribution before compensation.One of the advantages of smFRET measurement is the ability to trace the time evolution of the FRET changes in realtime like shown in Figures 2. If the FRET efficiency varies with changes in the interdye distance, the acceptor intensity increases while the donor intensity decrease, or vice versa (Figure 2A). However, the fluorescence intensity can be modulated by other reasons than FRET, such as photochemical effects like blinking or photobleach, which are explicitly observed in the single-molecule measurements. When such fluctuations occur, the anti-correlation relationship of the fluorescence intensities is diminished (Figure 2B). In order to distinguish the intensity modulation due to the FRET changes from those instabilities, calculating the cross correlation between two intensity signals was proposed to quantitatively evaluate the anti-correlation relationship[31]. Or more simply, the compensated total intensity Ic can be calculated as[30] 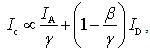
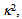 which depends on the relative orientation of two dyes while it is generally difficult to know the actual molecular orientation in real experiments. However, when dyes are labeled via linkers,
which depends on the relative orientation of two dyes while it is generally difficult to know the actual molecular orientation in real experiments. However, when dyes are labeled via linkers, 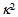 may be approximated to 2/3, which is the theoretical average under the assumption of random orientation. If one can obtain EFRET values for three different FRET states, the relative distance change can be absolutely calculated.The FRET efficiency can be measured from the fluorescence lifetime, too. The fluorescence lifetime is the time lag between the absorption of the excitation light and the fluorescence photon emission on a fluorophore. In order to measure the fluorescence lifetime, which is typically in the order of nanoseconds, a pulsed laser is used for excitation and the decay constant for an exponential distribution of the lag times is calculated from a bunch of photon detection. Then the FRET efficiency can be obtained as
may be approximated to 2/3, which is the theoretical average under the assumption of random orientation. If one can obtain EFRET values for three different FRET states, the relative distance change can be absolutely calculated.The FRET efficiency can be measured from the fluorescence lifetime, too. The fluorescence lifetime is the time lag between the absorption of the excitation light and the fluorescence photon emission on a fluorophore. In order to measure the fluorescence lifetime, which is typically in the order of nanoseconds, a pulsed laser is used for excitation and the decay constant for an exponential distribution of the lag times is calculated from a bunch of photon detection. Then the FRET efficiency can be obtained as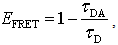
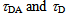 are the fluorescence lifetime of the donor dye with and without FRET, respectively[28].
are the fluorescence lifetime of the donor dye with and without FRET, respectively[28].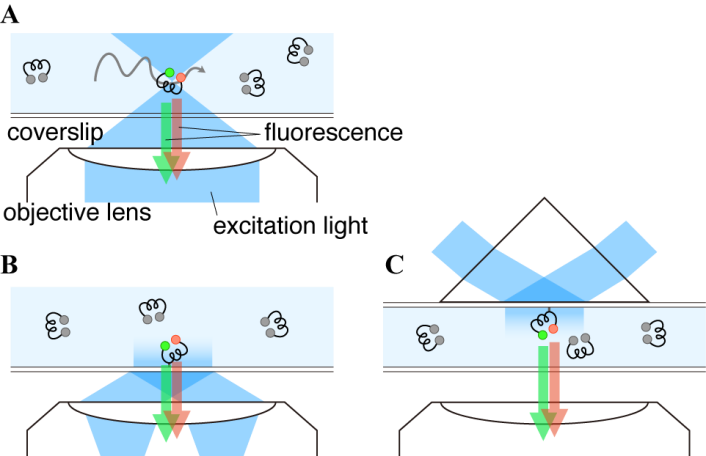
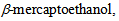 to suppress the fluorescence instability or the oxygen scavenger, such as the combination of glucose, glucose oxidase and catalase, to suppress the photobleach, are often mixed into the solution.
to suppress the fluorescence instability or the oxygen scavenger, such as the combination of glucose, glucose oxidase and catalase, to suppress the photobleach, are often mixed into the solution.