-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Physical Chemistry
p-ISSN: 2167-7042 e-ISSN: 2167-7069
2015; 5(2): 23-33
doi:10.5923/j.pc.20150502.01
A Quantitative and Qualitative Investigation of Conformational Analysis, Tautomeric Preference and Intramolecular Hydrogen Bonding on N-nitroso N-thionitrosamine (NTA)
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLPeyman Mohammadzadeh Jahani1, Nosrat Madadi Mahani2
1Bam University of Medical Sciences, Bam, Iran
2Department of Chemistry, Payam Noor University, Tehran, Iran
Correspondence to: Nosrat Madadi Mahani, Department of Chemistry, Payam Noor University, Tehran, Iran.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Various aspects of NTA were performed using B3LYP and MP2 methods with 6-311++G (d,p) basis set. First, the conformational analysis and characterization of equilibrium conformations, especially global minima, were carried out. On the basis of relative energies, we found that the N-nitroso N-thionitroso amine (TN) tautomer is more stable than the others, which is followed by1-hydroxy 2-thionitroso diazene (HT) and 2-mercpto diazene (NM) tautomers. This preference is well interpreted in terms of tautomerization process, thionitrosamine resonance and charge transfer energies. Furthermore, the nature of S-H  O and O-H…S intramolecular hydrogen bond (IMHB), in chelated forms of HT-11, NM-11, HT-13 and NM-13 were comprehensively studied to evaluate the effect of hetero atoms N and S on the characteristic of intramolecular hydrogen bond systems. The results of isodesmic method clearly show that this approach is unsuitable for estimation the IMHB energy. Whereas, both of the energy–geometry correlation and the electron density analysis Espinosa emphasize on HT-11 as a chelated form with the strongest hydrogen bond, that is followed by MA, NM-11, HT-13 and NM-13. Additionally, the geometrical, atoms in molecules (AIM) and natural bond orbital's (NBO) parameters also are in line with this conclusion.
O and O-H…S intramolecular hydrogen bond (IMHB), in chelated forms of HT-11, NM-11, HT-13 and NM-13 were comprehensively studied to evaluate the effect of hetero atoms N and S on the characteristic of intramolecular hydrogen bond systems. The results of isodesmic method clearly show that this approach is unsuitable for estimation the IMHB energy. Whereas, both of the energy–geometry correlation and the electron density analysis Espinosa emphasize on HT-11 as a chelated form with the strongest hydrogen bond, that is followed by MA, NM-11, HT-13 and NM-13. Additionally, the geometrical, atoms in molecules (AIM) and natural bond orbital's (NBO) parameters also are in line with this conclusion.
Keywords: N-Nitroso compounds, Intramolecular hydrogen bond, Conformational Analysis, AIM and NBO
Cite this paper: Peyman Mohammadzadeh Jahani, Nosrat Madadi Mahani, A Quantitative and Qualitative Investigation of Conformational Analysis, Tautomeric Preference and Intramolecular Hydrogen Bonding on N-nitroso N-thionitrosamine (NTA), Physical Chemistry, Vol. 5 No. 2, 2015, pp. 23-33. doi: 10.5923/j.pc.20150502.01.
Article Outline
1. Introduction
- N-Nitroso compounds, which include nitrosamines and its derivatives, have been known for more than 100 years, but nothing was known of their toxicological properties until 1937, when Freund [1] described a laboratory poisoning by nitrosodimethylamine. Then Barnes and Magee [2] in 1954 described a thorough toxicological examination of the compound in several species, in which liver and lung injury caused death. This culminated in a chronic toxicity test in rats, which resulted in a high incidence of animals with liver tumors within a year [3]. The parent nitrosamine, NH2-NO, is of special interest in atmospheric chemistry and it has been studied in very much detail [4]. Several theoretical works have been carried out in nitrosamine and its derivatives including ab initio calculations relevant to the mechanism of chemical carcinogenesis by N-nitrosamines [5], investigation of the interaction of the nitrosamines with the active site of cytochrome P450 [6, 7], study of the electronic properties of the low-lying spin states of dimethylnitrosamine coordinated to Fe(Ш) heme models [8], studies of the origin of rotational barrier in nitrosamine, study of the kinetics parameters associated with the rotation of nitroso group about the N–N bond in five and six-membered cyclic nitrosamine compounds and analysis of rotational energy barrier in six-membered cyclic nitrosamine compounds [9-11]. Also, recently a theoretical work was carried out by our research group in dinitrosoamine [12].By replacement of a thionitroso functional group in dinitrosamine instead nitroso group, N-nitroso N-thionitros amine (NTA) is formed. This compound can participate in the N-nitroso N-thionitroso amine (TN) ↔ 1-hydroxy 2-thionitroso diazene (HT) ↔1-nitroso 2-mercpto diazene (NM) tautomeric equilibriums (Fig.1). Therefore, NTA can have three different tautomers which are converted to each other by tautomeric equilibrium. In HT and NM tautomers, the appropriate arrangement of functional groups can forms S-H…O and O-H…S intramolecular hydrogen bonds (IMHB) which are assisted by π-electrons delocalization. This kind of hydrogen bond is represented by RAHB symbol that links the strength of the hydrogen bond to the resonance in chelated rings [13]. The chelated enol forms of β-dicarbonyl compounds, especially malonaldehyde (MA) and its derivatives, are the simplest members of RAHB systems which have been extensively studied [14-17]. Similar to MA and dinitrosamine [12], NTA shows several interesting features, such as equilibriums tautomerism, nitrosamine and thionitrosamine resonance and S-H…O and O-H…S intramolecular hydrogen bonds. Moreover, the presence of three hetero atoms (N) and one hetero atom (S) have an important role in determining the molecular structure of equilibrium conformations, tautomeric preference, intramolecular interactions and π-electron delocalization. In spite of these attractive properties, as far as we know, no extensive theoretical study about the NTA was published. Consequently, we think that it is necessary to carry out a comprehensive theoretical study about various aspects of NTA.
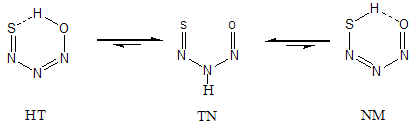 | Figure 1. Tautomeric equilibriums in NTA |
2. Computational Methods
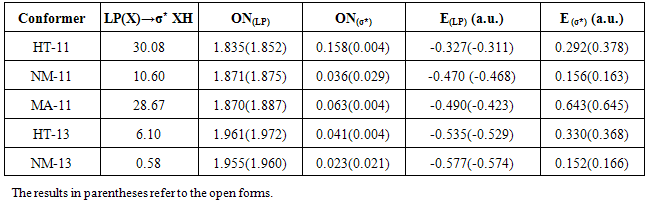
- All of calculations in this study were performed by GAMESS software ab initio package [18]. The geometry optimizations were carried out by B3LYP and MP2 methods with the standard, 6-311++G (d,p), basis set. Harmonic vibrational frequencies were evaluated at the same levels in order to identify the nature of the stationary points and employing the zero point vibrational energy (ZPVE) corrections. The optimized structures at B3LYP/6-311++G (d,p) level of theory were used to obtain the appropriate wave functions files for AIM and NBO analyses. The nature of the IMHB in the chelated forms has been studied using the AIM theory with AIM2000 software [19].The natural bond orbital analysis was carried out using the version 5.0 of NBO package [20] included in GAMESS program. NBO analysis stresses the role of orbital's interactions in studded molecule, particularly charge transfers. This is carried out by considering all the possible interactions between filled donor and empty acceptor NBOs and estimating their energetic importance by second-order perturbation theory. For each donor NBO (i) and acceptor NBO (j), the stabilization energy E(2) associated with electron delocalization between donor and acceptor is estimated as eq. (1):
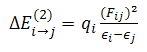 | (1) |
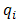 is the orbital occupancy,
is the orbital occupancy, 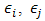 are diagonal elements and
are diagonal elements and 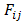 is the off-diagonal NBO Fock matrix element.Moreover, we estimated the IMHB energy of NM-11, NM-13, HT-11, HT-13 and MA according to the following methods:1. Electron density analysis of Espinosa. In this method, the IMHB energy is equal to half of the local potential energy density at critical point of hydrogen bond [21].2. Energy-geometry correlation of Musin and Mariam. In this method, the IMHB energy is determined based on the following eq. (2) [22].
is the off-diagonal NBO Fock matrix element.Moreover, we estimated the IMHB energy of NM-11, NM-13, HT-11, HT-13 and MA according to the following methods:1. Electron density analysis of Espinosa. In this method, the IMHB energy is equal to half of the local potential energy density at critical point of hydrogen bond [21].2. Energy-geometry correlation of Musin and Mariam. In this method, the IMHB energy is determined based on the following eq. (2) [22].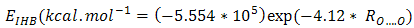 | (2) |
3. Results and Discussion
3.1. Conformational Analysis
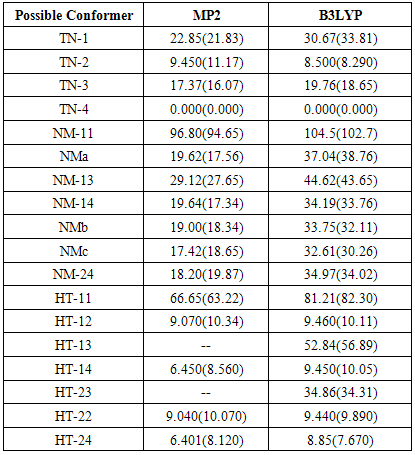
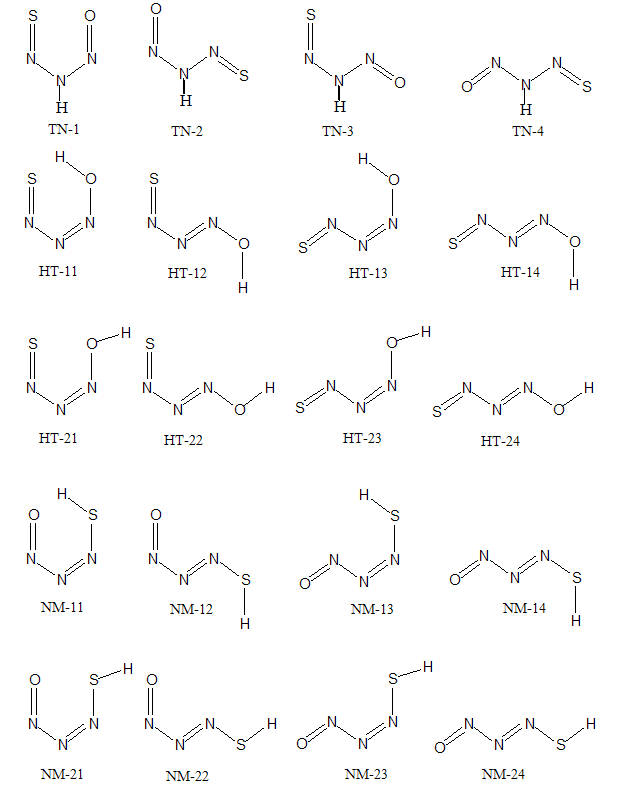
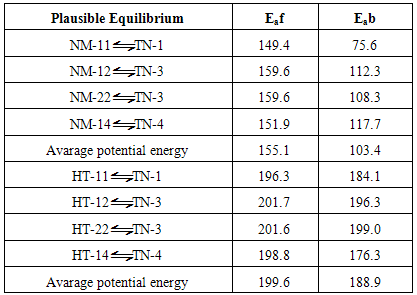
- Theoretically, NTA has about 20 plausible conformers which systematically arranged in three tautomeric classes, N-nitroso N-thionitroso amine (TN), 1-hydroxy 2-thionitroso diazene (HT) and 1-nitroso 2-mercpto diazene (NM) with 4, 8 and 8 members, respectively.Theoretical structures of these conformers and their relative energies are shown in Fig. 2 and Table 1. Based on relative energies, at all of the computational levels, it can be concluded that the energy of TN tautomer is slightly higher than the HT and NM tautomers. The investigation of conformational preference is the main goal of this section.
|
3.1.1. TN Tautomer
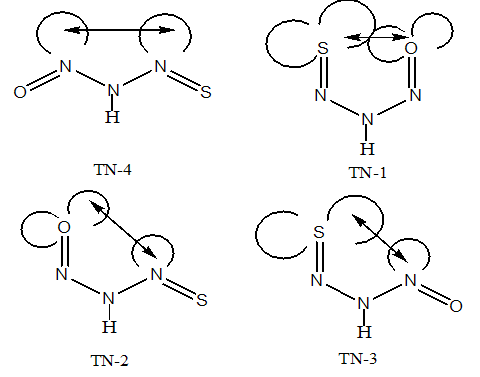
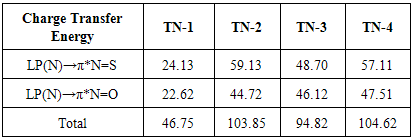
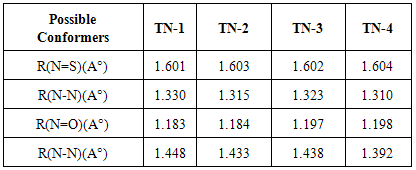
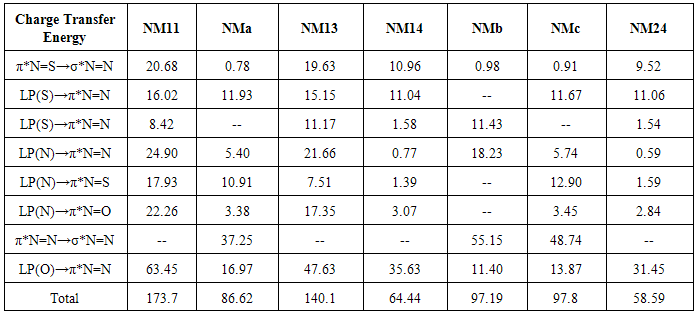
- From theoretical point of view, we considered four planar conformers for TN tautomer (TN-1, TN-2, TN-3 and TN-4), which are different in orientation of nitroso and thionitorso functional groups (Fig. 2). Full optimization and frequency calculation of TN forms, at different computational levels, shows four local minimum on the potential energy surface. Moreover, from the relative energies (Table 1), it can be concluded that the TN-4 is not only the most stable form among the TN conformers, which is followed by TN-2 , TN-3 and TN-1, but also is global minima amongst the known local minimums. It seems that the energy gap between the consecutive forms can be related to electrostatic repulsion amongst the nitrogen, oxygen and sulfur electron lone pair’s in nitroso and thionitroso functional groups and donor-acceptor stabilization charge transfers. In order to investigation of the electrostatic repulsion effects on the stability of TN forms, orientations of nitroso and thionitroso functional groups have been considered. In more stable form TN-4, the trans orientation of groups with respect to each other decrease the electrostatic repulsion while in more unstable form TN-1 the groups are in axial situation, which this configuration considerably increase the electrostatic repulsion. That‘s shown in Fig.3. In the other forms, TN-2 and TN-3 nitroso and thionitorso functional groups are in axial situation, respectively (Fig.3). It can conclude from Fig.3 which probably more polarizability of sulfur heteroatom in the thionitroso group increase overlap between electrons lone pairs sulfur and nitrogen atoms and unstable T-3 with respect to TN-2. These results are in good agreement with the energy order of TN conformers. The selected donor–acceptor charge transfer energy of TN forms, at B3LYP/6-311G (d,p) level of theory, are gathered in Table 2. The total values of charge transfer energies between the nitrogen lone pair and anti-bonding orbital of nitroso group (π*N=O) (nitrosamine resonance) and thionitorso group (π*N=S) (thionitosamine resonance) of TN-1, TN-2, TN-3 and TN-4 are about the 46.75, 103.85, 94.82 and 104.62kcalmol-1, respectively. These results are in good agreement with the energy order of TN conformers. Furthermore, the data in Table 2 clearly show that charge transfer energy between the central nitrogen lone pair and π*N=S is more in compare with π*N=O. This result refers to significant role of the high polarizability of sulfur heteroatom in electron delocalization. The nitrosamine and thionitosamine resonance significantly strengthened the N-N bonds of TN forms. As evident from Table 3, the N-N bonds lengths of TN-1, TN-2, TN-3 and TN-4, are about 1.330, 1.315, 1.323 and1.310 Å, respectively, which is in line with the previous conclusion.
 | Figure 2. The theoretical structure of possible conformations of the NTA |
 | Figure 3. Electrostatic repulsion between nitroso and thionitroso groups in TN conformers |
|
|
3.1.2. NM Tautomer
- Due to the presence of π-electron delocalization, eight planar NM conformers were proposed (Fig. 2). All of these theoretical forms were fully optimized by MP2 and B3LYP methods with standard basis set, 6-311++G (d,p). The absence of imaginary frequency reveals that all of these planar forms, except NM-12, NM-21, NM-22 and NM-23, have a particular local minimum on the potential energy surface of molecule. After full optimization, NM-12 changes to NMa and NM-22 changes to NMb while NM-21 and NM-23 convert to NMc. The optimized structures of NMa, NMb and NMc are presented in Fig. 4. During the optimization, the nitroso groups of NM-12, NM-22 and (NM-21, NM-23) pair have been rotated around the N-N bond by 72.89°, 95.50°, and 66.10°, respectively. These rotations may be assigned to the electrostatic repulsion between the oxygen, nitrogen and sulfur electrons lone pairs. The eclipsed orientation of lone pairs in the theoretical planar structures increases the electrostatic repulsion. But, the orientation of lone pairs in the optimized structures is staggered, which considerably reduce the electrostatic repulsion. The energy gaps between the initial and final structures reveal the difference of electrostatic repulsion between the eclipsed and staggered orientations of lone pairs. The corresponding values of NM-12, NM-22, and NM-21 and NM-23 pair are about 31.76, 36.86, 73.50 and 40.12 kJmol-1, respectively. Surprisingly, NM-11 with S-H
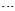 O intramolecular hydrogen bond is the least stable form. Also, there are significant energy difference between the least stable NM-11 form and other conformers. This instability is mainly due to the eclipsed orientation of five NM-11 lone pairs, which are maximized the repulsions. Furthermore, all of the computational levels emphasize on NMc as the most stable form whereas the relative energies of NMa, NMb and NM-24 are close or nearly degenerate (Table 2).
O intramolecular hydrogen bond is the least stable form. Also, there are significant energy difference between the least stable NM-11 form and other conformers. This instability is mainly due to the eclipsed orientation of five NM-11 lone pairs, which are maximized the repulsions. Furthermore, all of the computational levels emphasize on NMc as the most stable form whereas the relative energies of NMa, NMb and NM-24 are close or nearly degenerate (Table 2). | Figure 4. The optimized structures of NMa, NMb and NMc conformers |
 | Figure 5. The eclipsed and staggered orientation of lone pairs in some of NM conformers |
|
|
3.1.3. HT Tautomer
- Similarly of NM conformers, eight planar HT conformers were proposed (Fig. 2). All of these theoretical forms were fully optimized by MP2 and B3LYP methods with standard basis set, 6-311++G (d,p). The absence of imaginary frequency reveals that all of these planar forms have a particular local minimum on the potential energy surface of molecule. After full optimization, HT-21 converts to HT-23. During the optimization, the thionitroso group of HT-21 has been rotated around the N-N bond by 118.7°. This rotation may be assigned to the electrostatic repulsion between the nitrogen-nitrogen electron lone pairs that shown in Fig.6. The energy gaps between the HT-21 and HT-23 structures reveal the difference of electrostatic repulsion between the nitrogen-nitrogen electron lone pairs. The corresponding value is about 11.23 kJmol-1.
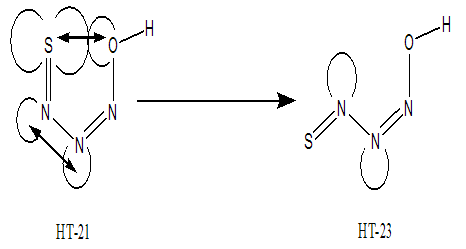 | Figure 6. Electrostatic repulsion between nitrogen-nitrogen lone pairs in HT-21 and HT-23 |
 S intramolecular hydrogen bond is the least stable form that after full optimization converts to HT-13. Also, HT-13 with O-H
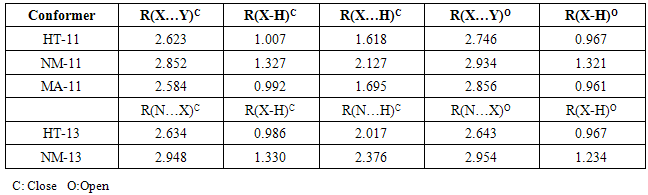
S intramolecular hydrogen bond is the least stable form that after full optimization converts to HT-13. Also, HT-13 with O-H N hydrogen bond, in a five membered ring, is not among the most stable forms. There are significant energy difference between the least stable (HT-11, HT-13) pair and other conformers. This instability is mainly due to the eclipsed orientation of five (HT-11) and four (HT-13) lone pairs, which are maximized the repulsions. Furthermore, all of the computational levels emphasizes on HT-24 as the most stable form whereas the relative energies of HT-14 and HT-24 are close or nearly degenerate. It seems that the greater stability of HT-14 and HT-24, with respect to other HT forms, is related to the minimization of electrostatic repulsions, owing to the staggered direction of lone pairs.The energy gap between HT-11 and HT-13, which differ only in ωN=N−N=S dihedral, reveals the repulsion between the nitrogen-nitrogen lone-pairs. This repulsion energy at B3LYP/6-311++G (d,p) level is about 28.37kJ mol-1 while these repulsion energy is lower than the corresponding value of dinitrosoamine(NH-11 and NH-13), 36.42kJ mol-1 [12]. Finally, the total values of charge transfer energies of HT-14, HT-24, HT-11and HT-13, are about 76.92, 84.46, 143.23 and 114.79 kcalmol-1, respectively. These results clearly show that the electrostatic repulsion is a superior factor in the relative stability of the HT forms.
N hydrogen bond, in a five membered ring, is not among the most stable forms. There are significant energy difference between the least stable (HT-11, HT-13) pair and other conformers. This instability is mainly due to the eclipsed orientation of five (HT-11) and four (HT-13) lone pairs, which are maximized the repulsions. Furthermore, all of the computational levels emphasizes on HT-24 as the most stable form whereas the relative energies of HT-14 and HT-24 are close or nearly degenerate. It seems that the greater stability of HT-14 and HT-24, with respect to other HT forms, is related to the minimization of electrostatic repulsions, owing to the staggered direction of lone pairs.The energy gap between HT-11 and HT-13, which differ only in ωN=N−N=S dihedral, reveals the repulsion between the nitrogen-nitrogen lone-pairs. This repulsion energy at B3LYP/6-311++G (d,p) level is about 28.37kJ mol-1 while these repulsion energy is lower than the corresponding value of dinitrosoamine(NH-11 and NH-13), 36.42kJ mol-1 [12]. Finally, the total values of charge transfer energies of HT-14, HT-24, HT-11and HT-13, are about 76.92, 84.46, 143.23 and 114.79 kcalmol-1, respectively. These results clearly show that the electrostatic repulsion is a superior factor in the relative stability of the HT forms.3.2. Tautomeric Preference
- Up to this point, we only identified the equilibrium conformations of tautomers and examined their preferences. These tautomers (TN, NM and HT) can be converted to each other by tautomeric equilibrium (Fig. 1). As previous mentioned, TN tautomer is slightly stable than the others, which is followed by HT and NM tautomers. This energy gaps can be related to the various factors such as tautomerization process, nitrosamine and thionitrosamine resonance of TN tautomer and π-electron delocalization of NM and HT tautomers.Enolization of keto functional group destabilize the molecule, because C=O, C-C and C-H bonds are broken and C-O, C=C and O-H bonds are formed. Based on bond average energies, the energy difference between the keto and enol forms of acetaldehyde is about 56 kJ mol-1, which is near to the computational energy gap [24]. By quantum chemical calculation at MP2/6-311++G (d,p) level we approximately obtained the same value [25]. In the same manner, it can be estimated the energy difference between the (nitrosamine, hydroxy-diazene) and (thionitrosamine, 1-mercaptodiazene) tautomers of the parent nitrosamine and thionitrosamine molecules (Fig. 7). From the average bond energies, this energy gaps between two tautomeric processes were estimated to be about 73 and 78kJ mol-1, respectively. These values show the energy gaps among of the TN, NM, and HT tautomers. Our computational results, at B3LYP/6-311++G (d,p) level, are in agreement with these conclusions. However, the theoretical energy gap between these tautomers is about 12.3 and 26.6 kJ mol-1, which is considerably lower than the previous values, 73 and 78 kJ mol-1. Probably, these differences are mainly due to the especial donor-acceptor charge transfers, which is usually, entitle as nitrosamine and thionitrosamine resonance. Similar to amide resonance, the nitrosamine and thionitrosamine resonance are ones of the most important stabilizing factors in TN tautomer with respect to NM and HT tautomers. In TN forms, the central nitrogen heteroatom can delocalize the electrons of its lone pair into the π* antibonding orbital's of nitroso and thionitroso functional groups. The most obvious feature of this phenomenon is lengthening of the N=O and N=S bonds, shortening of the N-N bonds, coplanarity of attached atoms, restricted rotation around the N-N bond and red shift nitroso and thionitroso stretching frequency. For instance, the (N=O, N=S) bond length of TN-1, TN-2, TN-3 and TN-4 are about (1.183, 1.601), (1.184, 1.603), (1.197, 1.602) and (1.198, 1.604) Å, respectively (see Table 3). It is apparent that these values are different from the corresponding N=O bond length of dinitrosamine with RN=O=1.208 Å and refer to significant role of sulfur heteroatom in more delocalize the central nitrogen electrons lone pair. [12] Hence, these results show that thionitrosamine resonance is higher than of nitrosamine resonance and can be considered as superior factor in the stability of TN forms.
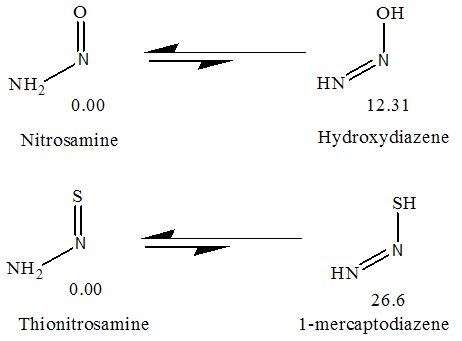 | Figure 7. The energy of tautomerism process in NTA |
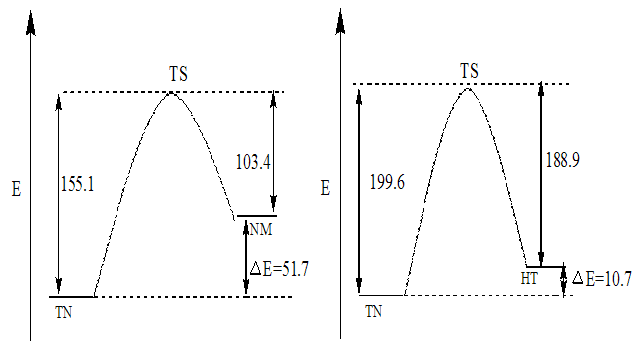 | Figure 8. The forward and backward potential energy of the barrier height of TN↔NM and TN↔ HT |
4. Hydrogen Bond
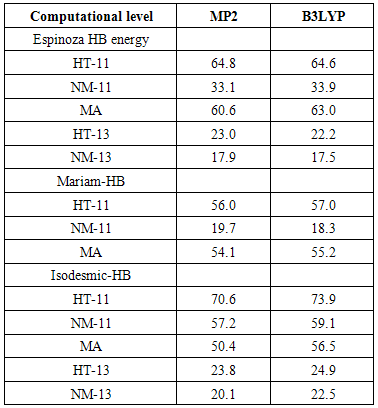
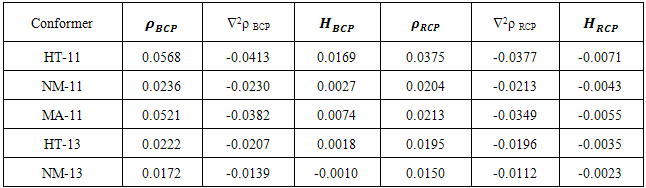
- Although, structures with the intramolecular hydrogen bond (IMHB) in five and six membered rings such as NM-11, NM-13, HT-11 and HT-13 are not present among the most stable forms, it is necessary to study the IMHB of them and evaluate the effect of hetero atoms N and S on the nature of hydrogen bond. Therefore, we estimated the IMHB energy of NM-11, NM-13, HT-11, HT-13 and MA by various models, such as electron density analysis of Espinosa, energy-geometry correlation of Musin and Mariam and isodesmic reactions method of Alkorta. Also, these results were compared with the results from dinitosamine molecule [12]. The calculated IMHB energies, at various computational levels, are gathered in Table 6. According to the results of isodesmic reaction method, the IMHB energy of NM-11 is greater than the MA. Whereas, both of the energy–geometry correlation and the electron density analysis Espinosa emphasize on HT-11 as a chelated form with the strongest hydrogen bond, that is followed by MA, NM-11, HT-13 and NM-13. In the isodesmic reactions method, the energy of the hydrogen bond is equal to the energy difference between the reactants and products of a suitable isodesmic reaction (Fig.9). However in products, a five membered ring with IMHB can be formed, which is shown by a dashed line that connects the H atom and NH2 group. Due to this interaction, the isodesmic approach is unsuitable for estimation the IMHB energy. Hence, the results of electron density analysis and energy–geometry correlation are more reliable. Consequently, the presence of three nitrogen heteroatom's (N) decreased the IMHB strength of NM-11 with respect to the chelated form of malonaldehyde, while the suitable position of sulfur atom in the HT-11 is a significant factor in its IMHB strength.
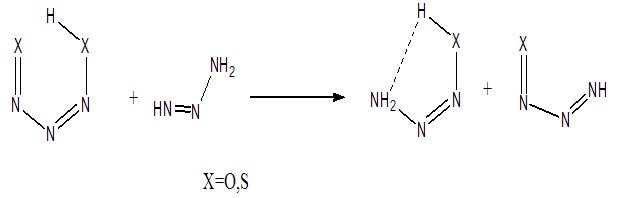 | Figure 9. Isodesmic reaction representation for estimate IMHB |
|
|
|
|
5. Conclusions
- Quantum chemical calculations, based on B3LYP and MP2 methods, are applied to study various aspects of potential energy surface of NTA. The results of ab-initio calculations were employed to evaluate the origin of conformational and tautomeric preferences. Additionally, the results of NBO and AIM analyses were used in order to estimate the IMHB strength. Following conclusions may be drawn from these theoretical calculations:1- According to vibrational frequencies calculations, NTA has about eighteen equilibrium conformers.2- At all of the theoretical levels, the TN forms have slight stability with respect to the other conformers. In addition, TN-4 form has greater stability with respect to the other forms and is global minima among the known local minimums.3- A detailed investigation of equilibrium conformations clearly show that the energy order of conformers is related to outcome influence of two superior factors, the electrostatic repulsion and charge transfer energies.4- A detailed investigation of tautomeric equilibrium clearly shows that three factors, tautomerism, thionitrosamine resonance and charge transfer energy can be rationalized the energy gaps of TN, NM and HT tautomers.5- According to the results of isodesmic method, it can be concluded that this approach is unsuitable for estimation the IMHB energy. For instance, based on the isodesmic method the IMHB energy of NM-11 is greater than the MA. Whereas, the energy-geometry correlation method, the electron density analysis, geometrical, topological and molecular orbital descriptors suggest that the HT-11 has the strongest IMHB, which is followed by MA, NM-11, HT-13 and NM-13.
ACKNOWLEDGEMENTS
- We gratefully thanks Bam University of medical science and Payam Noor University for financial support.