-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Physical Chemistry
p-ISSN: 2167-7042 e-ISSN: 2167-7069
2012; 2(1): 27-34
doi: 10.5923/j.pc.20120201.06
Heck Chemistry - a Highly Active Ligand-Free Metal Catalyst System in Situ Generated from PdII Supported on SiO2
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLLin Huang , Yu Wang , Zhan Wang , Fengxi Chen , Jozel Tan , Pui Kwan Wong
Heterogeneous Catalysis, Institute of Chemical and Engineering Sciences, A*STAR, 627833, Singapore
Correspondence to: Lin Huang , Heterogeneous Catalysis, Institute of Chemical and Engineering Sciences, A*STAR, 627833, Singapore.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
A ligand-free Pd catalyst system derived from Pd(acac)2/SiO2 and its catalytic properties toward the Heck coupling of bromobenzene and styrene have been studied. Combined studies by X-ray photoelectron spectroscopy (XPS), ultraviolet and visible absorption spectroscopy (UV-vis), thermogravimetric analysis (TGA) and powder X-ray diffraction (XRD) indicate that calcination of Pd(acac)2/SiO2 in air at 400℃ produces PdII/SiO2 that contains isolated Pd2+ ions coordinated to the lattice oxygen as the dominant component and PdO particles as the minor component. XPS and UV-vis observations together with catalytic results reveal that most of PdII is reduced to Pd0 on SiO2 by the reaction media during the Heck reaction and that the resulting catalyst system is highly active. Transmission electron microscopy measurements show that the in situ prepared Pd0/SiO2 has much finer Pd particles than conventional Pd0/SiO2. The rapid growth of the supported Pd particles during the highly efficient Heck reaction suggests that the catalysis occurs via an Ostwald ripening mechanism. Resulting in excellent catalytic properties, PdII/SiO2 possesses incomparable advantages over conventional Pd0/SiO2 as a precatalyst in catalytic activity, catalyst recycling and product contamination control.
Keywords: Heck reaction, in situ generated, palladium, silica, supported
Cite this paper: Lin Huang , Yu Wang , Zhan Wang , Fengxi Chen , Jozel Tan , Pui Kwan Wong , "Heck Chemistry - a Highly Active Ligand-Free Metal Catalyst System in Situ Generated from PdII Supported on SiO2", Physical Chemistry, Vol. 2 No. 1, 2012, pp. 27-34. doi: 10.5923/j.pc.20120201.06.
Article Outline
1. Introduction
- Among the various ways to synthesize arylated olefins, the construction of C(sp2)-C(sp2) single Bonds through Heck coupling is probably the most attractive way.1-5 It constitutes a very selective approach of forming new C-C bonds in a single operational step by coupling aryl and vinyl halides/triflates with olefins, in order to obtain a variety of substituted olefins, dienes and other unsaturated compounds.2,3,5Many of these compounds are useful as natural products, dyes, UV screens, raw materials for novel polymers, and intermediates for agrochemicals and pharmaceuticals.3,5 Significant benefits of developing Heck reactions lie in the tolerance of Heck reactions toward a large variety of functional groups on both reactants 1,5 and a wide availability of aryl chlorides and bromides. Traditional Heck reactions are typically carried out with 1-5 mol % of a Pd catalyst in the presence of a phosphine ligand and moreover the maximum turnover number (TON) attains 20-100 only.6 This obviously restricts large-scale industrial application. A major challenge in this area has in the past years been the development of new catalysts with higher TON. Quite a few advances have been made, including the use of sterically bulky and electron-rich phosphines, phosphorus-, nitrogen- and sulfur-based palladacyles and nucleophilic carbenes.6 However, all of these catalyst systems still suffer from drawbacks such as high ligand cost, high ligand sensitivity toward air and moisture, toxicity and the use of various additives. Ligand-free catalyst systems are viewed as promising for C-C coupling reactions from both operational and economic points of view.7-10 Since ligand-free Pd turned out to be feasible for Heck reactions on aryl iodides,11 extensive studies of ligand-free Pd catalyzed Heck reactions on aryl halides have increasingly been appearing.8,9,12-16 Due to the known drawbacks of homogeneous catalysts, it is desirable to develop heterogeneous Heck reaction catalysts for industrial applications and notable progress has been seen in this area.12-16 Instead of organic ligands, ligand-free Heck reaction catalyst systems usually employ a variety of inexpensive bases such as NaOAc, Na2CO3, NaHCO3, Ca(OH)2, Mg(OH)2, K3PO3, and amines.Amorphous SiO2 is one of the most commonly used supports for preparing heterogeneous metal catalysts. This material possesses the advantage of good stability in mechanics and chemistry. Nevertheless, little attention has been paid to on amorphous SiO2-supported metal catalyst systems for Heck reactions. To our knowledge, only a few limited catalytic results have appeared concerning Heck reactions over amorphous SiO2-supported Pd metal catalyst systems.17-26 Nearly all of these catalysts were conventionally prepared from PdII/SiO2 using a pre-reduction method. A conventional Pd0/SiO2 system has been considered to have no potential competitiveness with homogeneous Pd catalysts because its activity is far from that of homogeneous Pd catalysts. The only example of PdII/SiO2 used as a precatalyst has emerged in a letter about Heck couplings of aryl iodides and alkyl acrylates in the presence of Pd(NH3)4Cl2/SiO2.23 This supported complex was prepared by an ion-exchange method and was not subjected to any further treatments prior to Heck reactions. The catalytic activity was similar to that over conventional Pd0/SiO2 in an organic solvent, although it had a slight advantage in an ionic liquid.23
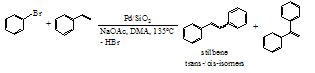 | Scheme 1. Heck coupling of PhBr and styrene with Pd/SiO2 |
2. Results and Discussion
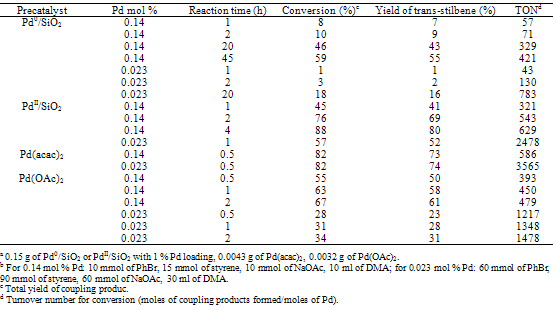
2.1. Behavior of Various Pd Catalysts in Heck Reactions
- Table 1 shows the comparative catalytic data of four different catalyst systems for the Heck coupling of PhBr and styrene at 135 oC. In the cases with 0.14 mol % Pd, Pd(acac)2 in solution resulted in the highest activity with a conversion of 82 % within 0.5 h; conventional Pd0/SiO2 displayed the lowest activity: The conversion was only 10 % within 2 h and did not reach a maximum of 59 % until after 45 h. The latter results are consistent with those reported for Pd0/SiO2 handled under similar conditions.21 Consistent comparative results were observed with 0.023 mol % Pd: Pd(acac)2 in solution was the most active system with a conversion of 82 % within 0.5 h; conventional Pd0/SiO2 was the least active with a conversion of 18 % at 20 h. Pd(OAc)2 in solution gave relevant results in accordance with those in the literature as well.6 Surprisingly, PdII/SiO2 brought about unusual activities: A conversion of 76 % was obtainable within 2 h and the reaction gave a maximal conversion of 88 % after 4 h with 0.14 mol % Pd; a conversion of 57 % was achievable within 1 h and the reaction presented a maximal conversion of 70 % with 0.023 mol % Pd. In fact, PdII/SiO2 gave rise to an activity intermediate between those of Pd(acac)2 and Pd(OAc)2 in solution. Our additional experimental data show that Pd(OAc)2, when utilized as a catalyst precursor to make Pd/SiO2, actually is not different from Pd(acac)2 in the catalytic properties of both conventional Pd0/SiO2 and PdII/SiO2 for this Heck reaction under the above working conditions.
|
|

 | Figure 1. Conversion of PhBr as a function of NaOAc:PhBr molar ratio in the Heck coupling of PhBr and styrene with PdII/SiO2 (0.14 mol % Pd) at 135℃ under Ar after 5 h |
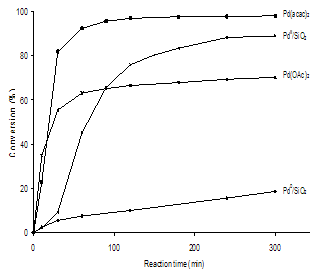 | Figure 2. Heck reaction profiles of PhBr and styrene with different catalyst systems (0.14 mol % Pd) at 135℃ under Ar |
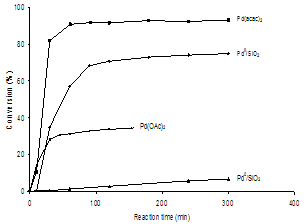 | Figure 3. Heck reaction profiles of PhBr and styrene with different catalyst systems (0.023 mol % Pd) at 135℃ under Ar |
|
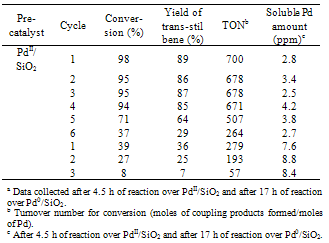
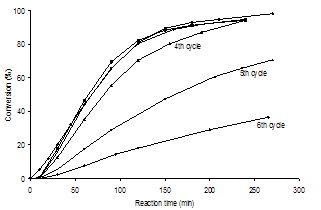 | Figure 4. Heck reaction profiles of PhBr and styrene with PdII/SiO2 (0.14 mol % Pd) at 135℃ under Ar |
2.2. Chemistry of PdII Supported on SiO2 with Regards to Heck Reactions
- The chemical evolution of Pd(acac)2-derived SiO2- supported samples with regards to a Heck reaction was probed with the aid of a variety of analytical methods. To our knowledge, a systematic monitoring of the evolution of a supported Pd precatalyst before and after a C-C coupling reaction has not yet been reported.Figure 5 presents the thermogravimetric analysis (TGA) profiles of Pd(acac)2 and Pd(acac)2/SiO2 having 1 % Pd loading under atmospheric air from room temperature to 400℃ at a heating rate of 10℃/min. The weight loss of Pd(acac)2/SiO2 below 180℃ is attributed to the desorption of molecular water adsorbed on the SiO2 surface. From 180℃, a second step of weight loss on Pd(acac)2/SiO2 consistently corresponds to the liberation of organic groups from Pd(acac)2, by comparison with the curve of weight loss of Pd(acac)2 alone. What is more, the overall TGA profile of Pd(acac)2/SiO2 clearly indicates that the thermal decomposition of Pd(acac)2 on SiO2 is already quantitatively complete in air by 250 oC. The markedly lower temperature (213 oC) at which Pd(acac)2/SiO2 decomposes completely than that of Pd(acac)2 (275 oC) alone implies that the interaction of Pd(acac)2 with SiO2 occurs significantly and speeds up the decomposition of Pd(acac)2, or that the size of Pd(acac)2 particles decreases upon deposition on SiO2. Figures 6 and 7 show the XPS spectra in the Pd 3d binding energy region and the UV-vis diffuse reflectance (UV-vis DR) spectra in the 200-800 nm region of Pd(acac)2 and Pd(acac)2-derived SiO2-supported samples, respectively. The XPS spectrum of Pd(acac)2 exhibited only a pair of intense Pd 3d3/2 and 3d5/2 peaks at 341.7 and 336.4 eV by deconvolution, which indicated the occurrence of a pure divalent Pd compound.37 However, the Pd 3d3/2 and 3d5/2 peaks of Pd(acac)2/SiO2 appeared at 340.5 and 335.2 eV. There was a marked downward shift of 1.2 eV in the Pd 3d binding energy after the deposition of Pd(acac)2 on SiO2 by impregnation. This change in XPS is in accordance with the difference between Pd(acac)2/SiO2 and Pd(acac)2 in TGA involving the interaction of Pd(acac)2 with SiO2. It may thus be assumed that some electron transfer from SiO2 to Pd as a result of the Pd(acac)2-SiO2 interaction is responsible for such a downward shift.
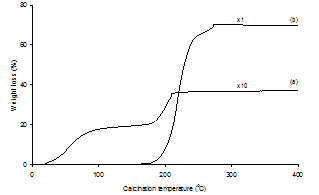 | Figure 5. TGA profiles in air from 22 to 400℃ at a heating rate of 10℃/min of (a) Pd(acac)2/SiO2 (1 % Pd loading); (b) Pd(acac)2 |
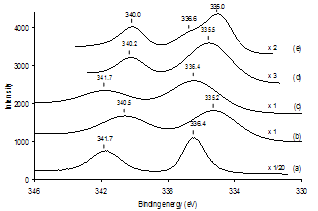 | Figure 6. Pd 3d3/2, 3d5/2 XPS spectra of (a) Pd(acac)2; (b) Pd(acac)2/SiO2; (c) Pd(acac)2/SiO2 after calcination in air at 400 oC; (d) Pd(acac)2/SiO2 after reduction in H2 at 400℃; (e) PdII/SiO2 after 2 h of Heck reaction |
 | Figure 7. UV-vis DR spectra of (a) Pd(acac)2; (b) Pd(acac)2/SiO2; (c) Pd(acac)2/SiO2 after calcination in air at 400 oC; (d) Pd(acac)2/SiO2 after reduction in H2 at 400℃; (e) PdII/SiO2 after 2 h of Heck reaction |
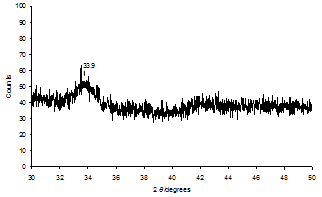 | Figure 8. XRD pattern of Pd(acac)2/SiO2 after calcination in air at 400℃ |
 | Figure 9. TEM images and Pd particle size distributions of (a) Pd0/SiO2 prepared by H2 reduction of Pd(acac)2/SiO2 at 400℃; (b) PdII/SiO2 after 2 h of Heck reaction at 135℃ and (c) PdII/SiO2 after the second Heck reaction cycle at 135℃ |
3. Conclusions
- By calcination in air at 400℃, Pd(acac)2/SiO2 is transformed into PdII/SiO2 consisting of isolated Pd2+ ions coordinated to lattice oxygen in majority and PdO particles in minority. Under the reaction conditions used here, most of the isolated surface Pd2+ ions and the PdO particles are reduced to the supported Pd particles by the reaction media. The resulting catalyst system turns out to be much more active for the Heck coupling of PhBr and styrene. The used catalyst can be recycled three times without significant loss of activity. The soluble Pd amount in the reaction mixture falls at an acceptable level ( < 5 ppm) after each reaction cycle. Pd0/SiO2 in situ generated in the Heck reaction at 135℃ has much finer Pd particles than Pd0/SiO2 conventionally prepared by H2 reduction at 400℃. The rapid growth of the supported Pd particles during the Heck reaction suggests that the supported smaller Pd particles are susceptible to leach molecular Pd into solution that takes part in the catalysis, after which molecular Pd aggregates onto the supported greater Pd particles. The in situ generated catalyst system possesses incomparable advantages over the conventionally prepared one in catalytic activity, catalyst recycling and product contamination control.
4. Experimental
- Silica gel (SiO2, Merck grade 10184) having a surface area of 300 m2/g, palladium acetylacetonate (Pd(acac)2, 99 %), palladium acetate (Pd(OAc)2, 99.8 %), PhBr (99 %), styrene (> 99 %), anhydrous sodium acetate (NaOAc, ≥ 99 %), anhydrous N,N-dimethylacetamide (DMA, 99.8 %) and anhydrous toluene (99.8 %) were purchased from Sigma-Aldrich. The gases H2 and Ar had a purity of 99.999 %.SiO2-supported Pd particle pre-catalysts were prepared as follows: SiO2 (1.0 g) was pre-dehydrated at 400℃ in flowing purified air for 8 h, and impregnated with a toluene (5 ml) solution of Pd(acac)2 (0.029 g) under Ar. The impregnated system was stirred for 2 h under Ar followed by evacuation of the solvent to give dry Pd(acac)2/SiO2 containing 1 % Pd. PdII/SiO2 and conventional Pd0/SiO2 were obtained from Pd(acac)2/SiO2 by calcination in flowing purified air at 400℃ for 8 h and by reduction in flowing H2 at 400℃ for 2 h, respectively. Before the treatments at 400℃, Pd(acac)2/SiO2 was heated from room temperature to 400℃ in flowing gas at a heating rate of 10℃/min in both cases.The catalytic reactions were conducted at 135℃ in glass flasks under Ar. In a typical experiment with 0.14 mol % Pd with respect to PhBr, to a 50 ml three-neck flask equipped with a septum were introduced 10 mmol of PhBr, 15 mmol of styrene, a certain amount of NaOAc, 0.15 g of heterogeneous precatalyst (Pd0/SiO2 or PdII/SiO2) or an equivalent amount of homogeneous precatalyst (Pd(acac)2 or Pd(OAc)2) under Ar. Then 10 ml of DMA was added as the solvent. After the mixture had been stirred at room temperature for 10 min under Ar, the flask was placed in a pre-heated oil bath with vigorous stirring (500 rpm). The reaction mixture was sampled at the reaction temperature and atmosphere through a 0.45 µm Whatman syringe filter. The reactants and products in the samples were analyzed by gas chromatography on a Perkin-Elmer Clarus 500 gas chromatograph with a J&W DB-1 capillary column (30 m x 0.320 mm x 1.00 µm) and a flame ionization detector. The Pd contents in the samples (leached Pd) were determined by the inductively coupled plasma (ICP) technique on a Varian Vista-MPX CCD simultaneous ICP-OES spectrograph.For the determination of the content of leached Pd, a certain volume of hot filtrate collected from the reaction mixture was first subjected to evaporation of the organic components in a glass vessel by heating under vacuum. Then a certain volume of aqua regia was added to the obtained sample. After gentle boiling for 10 min, the solution was diluted to a certain volume with deionized water followed by filtration with a Whatman syringe filter. The clear solution was analyzed by ICP. Analysis of each sample was performed twice regularly.For catalyst recycling, a solid sample containing a used supported catalyst was filtered off from the reaction mixture in air after a reaction cycle had ceased, and washed with DMA. Then it was directly transferred into a clean flask for the following reaction cycle.The oxidation states of Pd in Pd(acac)2 and Pd(acac)2- derived SiO2-supported samples were examined by XPS using an ESCALAB 250 X-ray photoelectron spectrometer with a non-monochromatic Al Kα radiation source at 1486.6eV and 200 W. Each sample was mounted on a sample holder with the aid of double-sided conducting adhesive tape. The sample was vacuum treated for 4 h before XPS analysis under 2.0 x 10-8 mbar. The UV-vis DR spectra of solid samples were recorded on a Shimadzu UV-2550 spectrophotometer. The phase structures of solid samples were determined by XRD on a Siemens D5005 spectrometer. The thermal decomposition of Pd(acac)2 and Pd(acac)2/SiO2 was monitored by TGA using an SDT 2960 simultaneous DSC-TGA analyzer. The microscopic images of supported Pd particles were observed by means of TEM on a JEOL TecnaiG2 microscope. The Pd particle size distributions were determined by counting the sizes of 100~150 Pd particles on several images taken from different places.
ACKNOWLEDGEMENTS
- This project was supported by funding from the Agency for Science, Technology and Research (A*STAR), Singapore.