-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Journal of Microbiology Research
p-ISSN: 2166-5885 e-ISSN: 2166-5931
2024; 14(1): 1-10
doi:10.5923/j.microbiology.20241401.01
Received: Mar. 15, 2024; Accepted: Apr. 7, 2024; Published: Apr. 13, 2024

Molecular Characterization of Heavy Metal Resistant Bacterial Isolates Obtained from Mining Soil in Ikpeshi, South-West, Nigeria
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML1Department of Microbiology, School of Applied Science and Technology, Auchi Polytechnic Auchi, Edo State, Nigeria
2Department of Microbiology, Adekunle Ajasin University, Akungba-Akoko, Ondo State, Nigeria; Centre for Infectious Disease Control and Drug Development (CIDCDD), Adekunle Ajasin University, Akungba-Akoko, Ondo State, Nigeria
Correspondence to: Ajayi A. O., Department of Microbiology, Adekunle Ajasin University, Akungba-Akoko, Ondo State, Nigeria; Centre for Infectious Disease Control and Drug Development (CIDCDD), Adekunle Ajasin University, Akungba-Akoko, Ondo State, Nigeria.
| Email: |  |
Copyright © 2024 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Mining activities have increased environmental pollution which has consequently resulted in the release of heavy metals into the environment. Bacteria are known to possess several detoxifying mechanisms to withstand the toxic effects of metal species in biotransformation and removal of these metals. The study shows the distribution of the indigenous metal species resistant bacteria in some abandoned mine effluents in Ikpeshi, Akoko-Edo, Nigeria. Soil samples were collected from Goopex quarry (S1), Freedom quarry (S2) Somak quarry (S3) and SC1, SC2, SC3 as their surrounding sites in Ikpeshi, Akoko-Edo, Nigeria. Bacterial isolates were identified by standard microbiological methods. Bacteria tolerance to higher concentration of these metals and their mechanisms of resistance were determined using molecular techniques. A total of 48 metal resistant bacteria species were isolated from three limestone mine tailing and soil in Ikpeshi, Akoko-Edo, Edo State, and using 1mM of Ni, Pb, Zn, Cd, Cr and Co. High resistance to Pb, Ni and Zn were observed in the isolated bacteria strains. Twenty-three of these isolates showed distinct morphological characteristics and the isolates showed tolerance to multiple metals at 5, 9 and 7 mM of Ni, Pb and Zn respectively. Ten isolates showing best growth were selected and their identity confirmed using 16S rRNA. Sequencing of the I6S rRNA gene and phylogenetic analysis of the nucleotide sequences determined from the 16S rRNA gene showed that these isolates belong to the genera Bacillus, Bacteriodes, Methanoococcus and Deferribacter, uncultured Sphingobacterium and uncultured Sulfuricurvum. Screening for the presence of plasmid revealed that the isolates were plasmid mediated. These results indicate that bacteria can be used for bioremediation of heavy metal contaminated site. Hence sampled sites need to be remediated. Measures should be taken to prevent water and wind erosion of the tailing to prevent further dispersal of metal species in the environment.
Keywords: Bacteria, Environmental pollution, Heavy metals, Metal species, Mine waste
Cite this paper: Yakubu P., Ajayi A. O., Molecular Characterization of Heavy Metal Resistant Bacterial Isolates Obtained from Mining Soil in Ikpeshi, South-West, Nigeria, Journal of Microbiology Research, Vol. 14 No. 1, 2024, pp. 1-10. doi: 10.5923/j.microbiology.20241401.01.
Article Outline
1. Introduction
- Mining activities have advanced the socio-economic development of national economies around the world; they have impacted negatively on the environment resulting in buildup of pollutants [1]. Incautious use of natural resources coupled with industrialization, mining, and urbanization has resulted in the buildup of pollutants in the environment [2,3]. This leads to great impact on ecosystem functioning because bacteria play a major role maintaining soil fertility and structure [4]. Bacteria are very sensitive and respond quickly to changes in environmental condition; they are therefore considered to be efficient bio-indicators of soil quality [5]. Bacteria in metals contaminated sites are able to overcome the stress imposed by metal species and thus make them to have a functional role in remediation of the polluted sites [6]. The stress imposed has led to the establishment of various defense mechanisms by the native bacteria community to tolerate the metals [7,8]. Numerous studies have shown that indigenous bacteria isolated from metal species contaminated sites effectively interact with toxic metals via direct and indirect mechanisms. The various resistance ability is a direct response to the particular metal species and consequently a given bacteria may directly and/ or indirectly rely on many survival mechanisms [9]. Due to this reason, bacteria are seen as tools for the treatment of metal species polluted sites in biological processes known as bioremediation [10]. They serve as alternative method to physico-chemical processes [11,12]. To effectively use these bacteria in bioremediation, it is compulsory to isolate, cultivate and select the desired strains because bacteria are known to be metabolically heterogeneous. Having a better knowledge of the mechanism controlling growth and activity of the bacteria in contaminated sites, their metabolic capabilities and response to environmental changes will also enhance their successful application. Despite large numbers of abandoned limestone mine tailings in Ikpeshi, Akoko Edo, Edo State, there is limited information on metal species resistant bacteria from the various hazardous wastes. Therefore, research on indigenous metal species resistant bacteria associated with mine tailings is important to know the diversity and physiology of metal species resistant bacteria that are thriving under unfavourable metal pollution. Hence, the aim of this research was to isolate metals resistant bacteria associated with limestone mine tailings and soils as well as identify and characterize them for their use as a prospective inoculant in bioremediation of metals polluted sites.
2. Methodology
- Soil sampling and analysisSoil samples were collected from active limestone mines in Ikpeshi, Akoko-Edo Edo State. Control soil samples were collected around the mines. Soil samples were collected in triplicate at depths of 10-30 cm with a soil auger and transported to the laboratory for analysis in sterile plastic bags.Preparation of stock solution of metals All glassware was autoclaved prior to use. The metal salts, ZnSO4, NiCl2.H2O, CoCl·6H2O, CrO4, CdSO4-2H2O and Pb(NO3)2 used were of analytical grade purchased from Sigma Aldrich, South Africa. One molar stock solutions of Pb, Ni, Zn, Cd and Co were prepared by dissolving ZnSO4, NiCl2.H2O, CoCl·6H2O, CrO4, CdSO4-2H2O and Pb(NO3)2 respectively in milli-Q grade deionized water and later filter sterilized using 0.20 μm pore size membrane filter. The glass wares used were treated in 2N HNO3 rinsed several times with distilled water before use to avoid metal contamination [13]. Isolation of metal resistant bacterial strains Metal tolerant bacteria were isolated from the samples using the spread plate method. One gram of duplicate composite samples was suspended in 9 mL of saline solution in distilled water and vortexed for 1-2 mms at room temperature. These were serially diluted (10-1 to I0-7) and aliquots 0.1 ml dilution from 10-4, 10-5 and 10·6 were spread with a glass rod over triplicate nutrient agar medium supplemented with I mM of ZnSO4, NiCl2.H2O, CoCl·6H2O, CrO4, CdSO4-2H2O and Pb(NO3)2 (Van Nostrand et al., 2007). To minimize metal ion complexation, the medium was adjusted to pH 7 using 1.0 N NaOH or 1.0 N HCI. Plates were incubated at 37°C and 25°C for 24 - 48 h. Growth was observed and morphologically distinct colonies were selected and purified in the same medium and growth conditions [14]. All cultures were stored at -80°C in nutrient broth with 20% glycerol for further studies. Determination of maximum tolerable metal concentration of the isolates Maximum tolerable concentration (MTC) for Pb, Cr, Zn, Ni, Cd and Co was determined by agar dilution method [15]. A log-phase culture of the isolates was spot inoculated onto Nutrient agar plates supplemented with increasing concentration of metals. The plates were incubated at 37°C and 25°C for 24 - 48 h and observed for bacterial growth. The highest concentration of the metal at which no bacteria growth was seen was designated as the MTC. All metal salts were added to the Nutrient agar after autoclaving and cooling to 50°C from filter-sterilized stock solutions. The concentrations in mM of the metal species used are stated below:
 Determination of multiple metal mixtures toleranceMulti elemental studies were done using combinations of two metals: Ni+Co, Ni+Pb and Zn+Pb. A log-phase culture of the isolates was inoculated into I0 μI of nutrient broth supplemented with lmM each of Ni, Pb, Zn and 0.5 mM of Co. The plates were incubated at 37°C and 25°C for 24 h-48 h and observed for bacterial growth by checking the bacterial OD at 600 nm using the UV spectrophotometer. All metal salts were added to the nutrient broth after autoclaving and cooling to 50°C from filter-sterilized stock solutions.Characterization and identification of the isolates Pure culture of bacteria isolates were subjected to various morphological, physiological and biochemical features e.g appearance and colour on agar medium, shape, size of colony, surface elevation, consistency, emulsifiability and gram staining. The result of each test was recorded and the probable identity to the isolates was deduced with references to Bergey’s Manual of Determinative bacteriology.Physiological characterizationIn selecting metal tolerant strains, the physiological conditions necessary for their growth was determined. The physiological conditions that are of great importance to bacteria growth are temperature, pH and salt (NaCl) concentration.Salt tolerance To determine salt tolerance of the isolate, 24 h pure cultures of each bacteria strains were streaked inoculated on Nutrient agar supplemented with I mM Pb and 1-I 0% NaCl which acts as a selective medium and Petri plates were incubated at 25°C and 37°C for 24 - 48 h. Bacteria were marked positive or negative for their ability to grow at different salt concentrations [16]. pH profileHydrogen ion concentration (pH) has a profound effect on bacteria growth. To determine optimal pH necessary for growth, Nutrient agar supplemented with 1 mM of Pb was used to grow the isolates. It was adjusted to different pH ranging from 3.0 - 9.0 using 1.0 N HCI and NaOH and then inoculated with the pure cultures of the isolates and the plates were incubated at 25°C and 37°C for 24 - 48 h. Growth was measured by the presence or absence of growth on the solidified agar medium. Temperature profileTo determine the optimum temperature, the pure isolates were inoculated into Nutrient agar plates supplemented with 1 mM of Pb. Overnight incubation was done at different temperatures; 25, 37, 40 and 50°C. The growth was measured by checking for the presence or absence of growth on the solidified agar plates. Molecular characterization of bacterial isolatesDNA EXTRACTIONGenomic DNA extraction Using DNA extraction kit from environmental sample (ABM Ontario Canada). Genomic DNA was extracted from the selected bacteria by growing in 10 ml of metal supplemented Nutrient broth. The bacterial cultures were grown for 24 h in a shaking incubator (150 rpm) maintained at 37°C and 1.5 ml of each culture was transferred to sterile Eppendorf tubes which was centrifuged at I000 rpm for 5 min. Supernatant were discarded and cells were re-suspended in 650 μI of TE buffer. Total genomic DNA was extracted from each bacterial suspension using a ZR soil microbe DNA mini prep TM DNA extraction kit (Zymo Research, USA) according to the manufacturer's protocol. Agarose gel electrophoresisThe presence of the genomic DNA and plasmid DNA was confirmed in a 1.0% (w/v) agarose gel electrophoresis prepared by dissolving 1.0 g of agarose (Bio-Rad, SA) in 100 ml of IX Tris-acetate-ethylenediaminetetraacetate (TAE, pH 8). The mixture was heated in a microwave oven for 3 mins and allowed to cool after which 10 μI of ethidium bromide (Bio-Rad, SA) was added to the molten agar which was poured in a gel casting tray and allowed to solidify. After solidification, the combs were removed and the gel was carefully placed in the electrophoresis tank containing 1 X TAE buffer (40 mM Tris, 20 mM Acetic acid, and 100 mM EDTA pH 8.0) . DNA samples were prepared by mixing 5μ1 of the genomic DNA with 5μ1 of 6X DNA loading dye (Thermo Scientific™) and this was carefully loaded in the pre-formed wells in the gel. A GeneRuler™ DNA Ladder (1 kb) was used to estimate the sizes of the genomic DNA. The electrophoresis was carried out at 100 V, 450 mA for 1 h. Gels were visualized and photographed using a gel documentation system (Gel Doc 2000, Bio-Rad). Polymerase chain reaction (PCR) amplification The 16S rRNA gene was amplified from genomic DNA obtained from bacterial cultures by PCR using universal bacterial I 6S rRNA primers 27F (5'-AGA GTT TGA TCC TGG CTC AG-3') and 1492R (5'-GGTTAC CTT GTT ACG ACT T-3') previously described by Liu et al. (2007). Polymerase chain reaction (PCR) was performed in a total volume of 50 μI 107 containing 25 μI of 2x PCR master mix (0.05 U/μI Taq DNA polymerase, 4 mM MgCb and 0.4 mM dNTPs) (Thermo Scientific™), 2 μI of genomic DNA template, 1.0 μI of each forward and reverse primer and 22 μI of nuclease free water. The amplification reaction mixture was subjected to 30 cycles in a C 1000 thermal cycler (BioRad, USA). The thermal cycling condition used was an initial denaturation, 95°C for 5 min; followed by denaturation at 95°C for 1 min; annealing, 58°C for 30s; extension, 72°C for I min and final extension, 72°C for 7 min. The PCR amp I icons were analyzed by electrophoresis in 1% (w/v) agarose gel and the sizes of the bands were determined using 1 kb molecular marker. The gel containing ethidium bromide (10μg/ml) were visualized and photographed using a gel documentation system (Gel Doc 2000, Bio-Rad) to confirm the expected size of the PCR products. PCR products were gel extracted (Zymo Research, Zymoclean™ Gel DNA Recovery Kit). Sequencing reaction The forward and reverse primers were both used in the sequencing of the purified PCR products. The sequencing was done at Inqaba Biotechnical industrial (Pty) Ltd, Pretoria, South Africa with PRISM TM Ready Reaction Dye Terminator Cycle Sequencing Kit using dideoxy chain termination method and electrophoresed with a model ABI PRISM® 3500XL DNA Sequencer (Applied Bio systems, USA) by following the manufacturer' s instructions. Purified sequencing products (Zymo Research, ZR-96 DNA Sequencing C lean-up Kit™) were analyzed using CLC Main Workbench 7. Phylogenetic analysisSequence similarities and phylogenetic analysis The chromatograms resulting from sequencing reaction were edited using Chromas Lite version 2.4 software [17]. The resulting nucleotide sequences were then analyzed and edited using Bio Edit Sequence Alignment Editor (Hall and CA, 2004). The consensus l6S rRNA sequences generated were compared with other reference sequences available in the National Center for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov) database using the Basic Alignment search tool (BLASTn) [18] and the sequences were deposited in the GenBank. Multiple alignment of the nucleotide sequences were done using Mafft version 7.0 [19]. Phylogenetic and molecular evolutionary analyzes were conducted using softwares in MEGA version 6.0 [20]. Evolutionary distance matrix was generated and a phylogenetic tree was drawn by neighbour joining method [21]. The I6S rRNA gene sequences of the selected metal resistant bacterial isolates determined in this study were deposited in NCBI/ EMBL nucleotide sequence GenBank database. Plasmid extractionPlasmid DNA was extracted by the alkaline lysis method as described by Kado [22]. Isolates was inoculated into nutrient broth and incubated for 24hrs at 37°C, after each bacterial culture was suspended in micro centrifuge with lysis buffer. It was heated for 15mins at 70°C and mixed with an equal volume of precooled 100% ethanol. The supernatant was discarded and the pellet was rinse twice with 1ml of 70% ethanol. It was air dried for at least 10mins and finally resuspended in 20μl of TBE (Tris-borate- EDTA) and store in the refrigerator for electrophoresis. The plasmid DNA was run in 1% agarose gel electrophoresis.
Determination of multiple metal mixtures toleranceMulti elemental studies were done using combinations of two metals: Ni+Co, Ni+Pb and Zn+Pb. A log-phase culture of the isolates was inoculated into I0 μI of nutrient broth supplemented with lmM each of Ni, Pb, Zn and 0.5 mM of Co. The plates were incubated at 37°C and 25°C for 24 h-48 h and observed for bacterial growth by checking the bacterial OD at 600 nm using the UV spectrophotometer. All metal salts were added to the nutrient broth after autoclaving and cooling to 50°C from filter-sterilized stock solutions.Characterization and identification of the isolates Pure culture of bacteria isolates were subjected to various morphological, physiological and biochemical features e.g appearance and colour on agar medium, shape, size of colony, surface elevation, consistency, emulsifiability and gram staining. The result of each test was recorded and the probable identity to the isolates was deduced with references to Bergey’s Manual of Determinative bacteriology.Physiological characterizationIn selecting metal tolerant strains, the physiological conditions necessary for their growth was determined. The physiological conditions that are of great importance to bacteria growth are temperature, pH and salt (NaCl) concentration.Salt tolerance To determine salt tolerance of the isolate, 24 h pure cultures of each bacteria strains were streaked inoculated on Nutrient agar supplemented with I mM Pb and 1-I 0% NaCl which acts as a selective medium and Petri plates were incubated at 25°C and 37°C for 24 - 48 h. Bacteria were marked positive or negative for their ability to grow at different salt concentrations [16]. pH profileHydrogen ion concentration (pH) has a profound effect on bacteria growth. To determine optimal pH necessary for growth, Nutrient agar supplemented with 1 mM of Pb was used to grow the isolates. It was adjusted to different pH ranging from 3.0 - 9.0 using 1.0 N HCI and NaOH and then inoculated with the pure cultures of the isolates and the plates were incubated at 25°C and 37°C for 24 - 48 h. Growth was measured by the presence or absence of growth on the solidified agar medium. Temperature profileTo determine the optimum temperature, the pure isolates were inoculated into Nutrient agar plates supplemented with 1 mM of Pb. Overnight incubation was done at different temperatures; 25, 37, 40 and 50°C. The growth was measured by checking for the presence or absence of growth on the solidified agar plates. Molecular characterization of bacterial isolatesDNA EXTRACTIONGenomic DNA extraction Using DNA extraction kit from environmental sample (ABM Ontario Canada). Genomic DNA was extracted from the selected bacteria by growing in 10 ml of metal supplemented Nutrient broth. The bacterial cultures were grown for 24 h in a shaking incubator (150 rpm) maintained at 37°C and 1.5 ml of each culture was transferred to sterile Eppendorf tubes which was centrifuged at I000 rpm for 5 min. Supernatant were discarded and cells were re-suspended in 650 μI of TE buffer. Total genomic DNA was extracted from each bacterial suspension using a ZR soil microbe DNA mini prep TM DNA extraction kit (Zymo Research, USA) according to the manufacturer's protocol. Agarose gel electrophoresisThe presence of the genomic DNA and plasmid DNA was confirmed in a 1.0% (w/v) agarose gel electrophoresis prepared by dissolving 1.0 g of agarose (Bio-Rad, SA) in 100 ml of IX Tris-acetate-ethylenediaminetetraacetate (TAE, pH 8). The mixture was heated in a microwave oven for 3 mins and allowed to cool after which 10 μI of ethidium bromide (Bio-Rad, SA) was added to the molten agar which was poured in a gel casting tray and allowed to solidify. After solidification, the combs were removed and the gel was carefully placed in the electrophoresis tank containing 1 X TAE buffer (40 mM Tris, 20 mM Acetic acid, and 100 mM EDTA pH 8.0) . DNA samples were prepared by mixing 5μ1 of the genomic DNA with 5μ1 of 6X DNA loading dye (Thermo Scientific™) and this was carefully loaded in the pre-formed wells in the gel. A GeneRuler™ DNA Ladder (1 kb) was used to estimate the sizes of the genomic DNA. The electrophoresis was carried out at 100 V, 450 mA for 1 h. Gels were visualized and photographed using a gel documentation system (Gel Doc 2000, Bio-Rad). Polymerase chain reaction (PCR) amplification The 16S rRNA gene was amplified from genomic DNA obtained from bacterial cultures by PCR using universal bacterial I 6S rRNA primers 27F (5'-AGA GTT TGA TCC TGG CTC AG-3') and 1492R (5'-GGTTAC CTT GTT ACG ACT T-3') previously described by Liu et al. (2007). Polymerase chain reaction (PCR) was performed in a total volume of 50 μI 107 containing 25 μI of 2x PCR master mix (0.05 U/μI Taq DNA polymerase, 4 mM MgCb and 0.4 mM dNTPs) (Thermo Scientific™), 2 μI of genomic DNA template, 1.0 μI of each forward and reverse primer and 22 μI of nuclease free water. The amplification reaction mixture was subjected to 30 cycles in a C 1000 thermal cycler (BioRad, USA). The thermal cycling condition used was an initial denaturation, 95°C for 5 min; followed by denaturation at 95°C for 1 min; annealing, 58°C for 30s; extension, 72°C for I min and final extension, 72°C for 7 min. The PCR amp I icons were analyzed by electrophoresis in 1% (w/v) agarose gel and the sizes of the bands were determined using 1 kb molecular marker. The gel containing ethidium bromide (10μg/ml) were visualized and photographed using a gel documentation system (Gel Doc 2000, Bio-Rad) to confirm the expected size of the PCR products. PCR products were gel extracted (Zymo Research, Zymoclean™ Gel DNA Recovery Kit). Sequencing reaction The forward and reverse primers were both used in the sequencing of the purified PCR products. The sequencing was done at Inqaba Biotechnical industrial (Pty) Ltd, Pretoria, South Africa with PRISM TM Ready Reaction Dye Terminator Cycle Sequencing Kit using dideoxy chain termination method and electrophoresed with a model ABI PRISM® 3500XL DNA Sequencer (Applied Bio systems, USA) by following the manufacturer' s instructions. Purified sequencing products (Zymo Research, ZR-96 DNA Sequencing C lean-up Kit™) were analyzed using CLC Main Workbench 7. Phylogenetic analysisSequence similarities and phylogenetic analysis The chromatograms resulting from sequencing reaction were edited using Chromas Lite version 2.4 software [17]. The resulting nucleotide sequences were then analyzed and edited using Bio Edit Sequence Alignment Editor (Hall and CA, 2004). The consensus l6S rRNA sequences generated were compared with other reference sequences available in the National Center for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov) database using the Basic Alignment search tool (BLASTn) [18] and the sequences were deposited in the GenBank. Multiple alignment of the nucleotide sequences were done using Mafft version 7.0 [19]. Phylogenetic and molecular evolutionary analyzes were conducted using softwares in MEGA version 6.0 [20]. Evolutionary distance matrix was generated and a phylogenetic tree was drawn by neighbour joining method [21]. The I6S rRNA gene sequences of the selected metal resistant bacterial isolates determined in this study were deposited in NCBI/ EMBL nucleotide sequence GenBank database. Plasmid extractionPlasmid DNA was extracted by the alkaline lysis method as described by Kado [22]. Isolates was inoculated into nutrient broth and incubated for 24hrs at 37°C, after each bacterial culture was suspended in micro centrifuge with lysis buffer. It was heated for 15mins at 70°C and mixed with an equal volume of precooled 100% ethanol. The supernatant was discarded and the pellet was rinse twice with 1ml of 70% ethanol. It was air dried for at least 10mins and finally resuspended in 20μl of TBE (Tris-borate- EDTA) and store in the refrigerator for electrophoresis. The plasmid DNA was run in 1% agarose gel electrophoresis. 3. Results
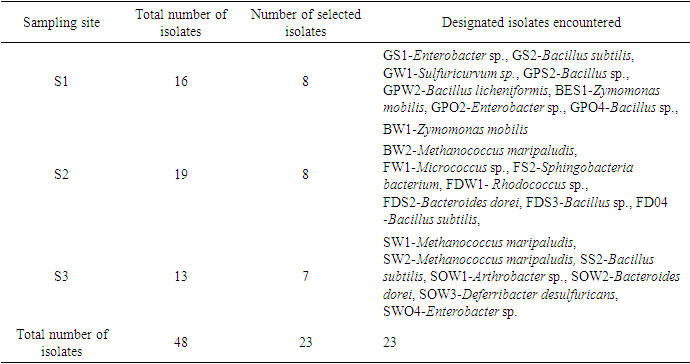
- A total of 48 culturable metal species resistant bacteria were isolated from the three sites S1, S2 and S3 under study using ImM concentration each of Pb, Zn, Ni, Co, Cr and Cd. Twenty-eight (23) of these isolates identified by standard microbiological methods clearly tabulated include GS1-Enterobacter sp., GS2-Bacillus subtilis, GW1-Sulfuricurvum sp., GPS2-Bacillus sp., GPW2-Bacillus licheniformis, BES1-Zymomonas mobilis, GPO2-Enterobacter sp., GPO4-Bacillus sp., BW1-Zymomonas mobilis, BW2-Methanococcus maripaludis, FW1-Micrococcus sp., FS2-Sphingobacteria bacterium, FDW1- Rhodococcus sp., FDS2-Bacteroides dorei, FDS3-Bacillus sp. and FD04 -Bacillus subtilis. other Bacterial isolates encountered were, SW1-Methanococcus maripaludis, SW2-Methanococcus maripaludis, SS2-Bacillus subtilis, SOW1-Arthrobacter sp., SOW2-Bacteroides dorei, SOW3-Deferribacter desulfuricans and SWO4-Enterobacter sp. (Table 1). In Table 2, most of the bacterial isolates tested against various metal species including heavy metals shows relatively high resistance at ImM concentration against Chromium (Cr2+), Lead (Pb2+) and Zinc (Zn2+). This ranged from average 6mM to 10mM for these metal species.
|
|

|

|
 | Plate 1. Fingerprint photograph showing amplicons of approximately 500bp for 16S rRNA gene amplification of the metal resistant bacteria |
 BACTERIA ISOLATES
BACTERIA ISOLATES
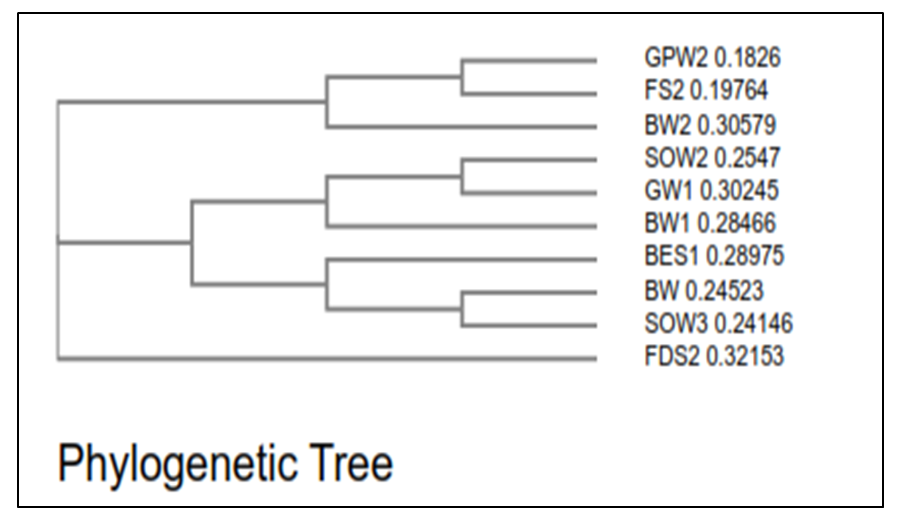 | Figure 1. Phylogenetic tree based on partial 16S rRNA gene sequence |
 | Plate 2. Plasmid profile |
4. Discussions
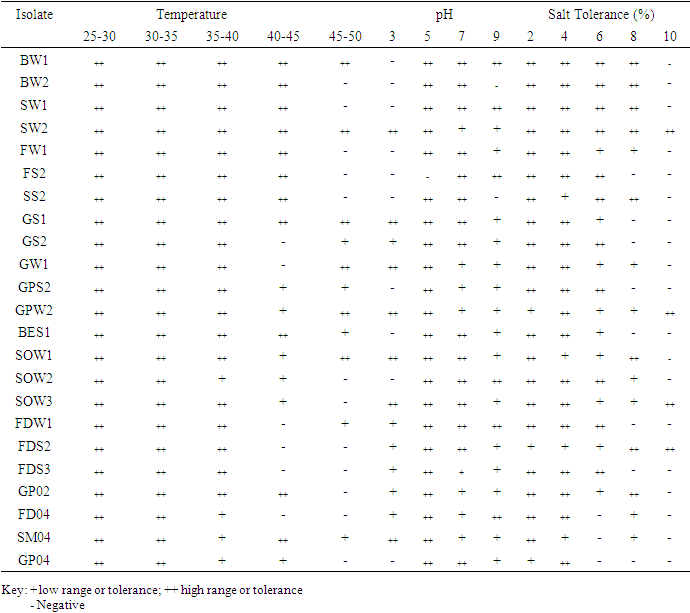
- A total of 48 culturable metal resistant bacteria were isolated from the three sites under study using ImM concentration each of Pb, Zn, Ni, Co, Cr and Cd. 23 of these isolates shows distinct colony characteristics like size, pigmentation, shape, elevation, and margin surface (Table 1). All the bacterial isolates tested could not grow beyond 1 mM of Co, but they were able to tolerate varying concentrations of Ni, Pb, Zn, Cd and Cr as shown in (Table 2). Cadmium tolerance range between 1-3 mM and five isolates; GPW2, BES1, BW1, SOW3 and GW1 could tolerate up to 3 mM concentration of this metal. Bacterial tolerance to Ni2+ range between 1-5 mM, isolates SOW2, FDS2, GPW2 and SOW3 shows highest tolerance of 5 mM to this metal. Tolerance to Pb also ranges between 1-7 mM for the isolates; SW2, GPW2 and SOW3 show the highest tolerance of 7 mM. Similarly, tolerance to Zn was also found to range between 1-9 mM and three isolates GPW2, SOW3 and FS2 were able to tolerate up to 9 mM concentration of this metal. Chromium was also well tolerated by the isolates; high tolerance of 7-10 mM was recorded. Isolates GPW2, SOW2, BW2 and SOW3 shows highest tolerance of 10 mM. Bacterial isolates GPW2 and SOW2 were observed to tolerate the highest concentration of 5 mM Ni, 7mM Pb, 9 mM Zn and 10 mM Cr2+ of the metal species tested. The high level of resistance and the widespread tolerance shown by the bacterial isolates against multiple metals could be attributed to the elevated concentrations of the metal species recorded in the different samples where the bacteria were isolated from. The differences in metal tolerance by the bacterial isolates could be attributed to many factors such as the medium strength, presence of negatively charged ions like chloride, organic constituents, and nature of the medium which determines availability of the metals to the bacteria (Kannan and Krishnamoorthy, 2006). The present study also shows that the bacterial isolates have multiple metal resistances (Table 3). The isolates show different tolerance to the multiple mixtures of Imetals tested. It was observed that Ni+Co combination was generally toxic to the bacterial isolates as observed in their reduced growth rate (Table 3). The bacterial isolates were able to tolerate the combination of Ni+ Pb and Zn+Pb better than Ni+Co. Out of the 23 isolates, ten isolates shows better growth in the presence of the multiple metal mixtures but the isolate GPW2 and SOW3 shows the best growth. All the isolates were further characterized for their physiological and biochemical characteristics to determine the probable identity of bacteria that could tolerate multiple metals in the mine tailings. The effect of temperature, pH and salt on bacteria growth in the presence of metal is shown in Table 4 The results shows that the optimum temperature for maximum growth by all the bacterial isolates was between 30°C-35°C, although majority of the isolates were also able to grow between 35°C-40°C. Decline in growth was observed when the temperature was raised to 40°C which could be due to the decrease in metabolic activity as a result of increase in temperature above the optimum value the bacteria can withstand. The observation corroborate with the studies of Adamo et al., [23] and Bajkic et al., [24], who reported that High temperature of growth medium affects the arrangement and stability of bacterial cell wall, ionized chemical moieties and also leads to disruption of the enzymatic activities which consequently slow down the metabolic activities of the bacteria. Similarly, low temperature also reduced bacterial growth because enzymes are inactivated at this temperature which decreases the rate of metabolism [25,26,27].The result of the effect of pH on bacterial growth indicated that optimum pH range for the bacterial growth is 5 and 7 while a decline in growth was observed at pH 3 and 9. The effect of salt on bacterial growth shows that increasing concentration of salt in the growth medium reduces the bacteria growth rate, maximum growth was observed at 2% followed by 4%. The result obtained is in accordance with the report of Fashola et al. [28] who showed that the bacteria are able to produce osmolytes like sugar and amino acids to protect themselves against the hypertonic environment created by the salt. At higher concentration of 6% -8%, growth was greatly reduced, some of the isolates could not withstand the high osmotic gradient created by salt again while at I0% only isolates SW2, GPW2, SOW3, and FDS2 could tolerate the high salt concentration in the growth medium (Table 4). The observation correlates with the study of Fashola et al. [28], who reported that effect of salt on bacterial growth shows that increasing concentration of salt in the growth medium reduces the bacteria growth.Ten isolates: GPW2, FS2, BW2, SOW2, GW1, BW1, BES1, SW2, SOW3 and FDS2 showed high metal tolerance. The amplification of the 16S rRNA genes from the genomic DNA yielded a 1.5 kb fragment as shown in (Plate 1). The BLAST query grouped the bacterial isolates into Bacillus, Bacteriodes, Methanoococcus, Deferribacter, Sphingobacterium and Sulfuricurvum sp.as shown in Fig 1. Exposure to metals has been reported to usually lead to tolerance in population of Gram positive and Gram negative bacteria present in contaminated sites. [29] as observed in this study. None of the isolates shows resistance to the metal species on the chromosomal DNA confirming the earlier claim that resistance to metal are majorly located on plasmid (Plate 2). These bacteria are known to apply various types of resistance mechanisms which could be plasmid or chromosomally encoded but these resistance has been mostly reported to be plasmid mediated [28,30,31]. In a study conducted by Raja and Selvam [14], resistance to Pb and Ni by Pseudomonas aeruginosa isolated from metal polluted waste water was found to be plasmid mediated. Similarly, Piotrowska-Seget et al. [32] also reported bacterial resistance to Zn in their study to be plasmid mediated. The presence of metal resistance determinants on plasmids has suggested that these genes may be transfered to divergent bacteria through horizontal gene transfer [33].
5. Conclusions
- This research invariably establishes some novel characteristics of isolates strains which had high level of adaptability to the mines environmental with biodegradable ability to degrade some heavy metals that are of high significance. This study is the first to report a heavy metal tolerance capacity of bacteria isolated from the soil of Ikpeshi, Akoko Edo, and mining areas. Studies undertaken to examine the identification and characteristics of environmental samples revealed the true diversity of microorganisms and their unique functionality which arise from their biological system that produce enzymes to make them tolerate or adapt to their environments. The use of molecular techniques adds more precision and accuracy to the phylogenetic identification and the true reflection of microbial diversity. Bacillus can be used for knowledge of the active microorganisms in the mines and is important for recovering metals and the development of optimal in situ bioremediation strategies. Many organisms encountered in this study shows significant growth at 48hours, exemplified by the optical density in multi-media cultures within the period of 24hours to 72hours. The growth range continues slowly amongst many of the organisms up to 72hours while few such SS2 -Bacillus subtilis and FDS3-Bacillus sp. decline for most of this metal mixture. Showing their low metal tolerance. That means, other bacterial isolates studied in this context can be used for bioremediation purposes which can safeguard the ecology and health of people in this are and human health generally.
6. Recommendations
- Measures should be taking to prevent water and wind erosion of the tailings as this will lead to further dispersal of metal species in the environment and the sampled sites need to be remediated urgently. More funding and support should also be provided by funding agencies, research development bodies and government to encourage implementation of bioremediation technology on a large scale as this hold promising future for the treatment of the enormous mine wastes generated by the country. Complementary to this further research can be done on the use of genetically modified organisms for this bioremediation purpose as well as the possibility of converting this waste sources for some valuable energy purpose in bid of making resource from the waste. Good database should be formulated for research development and health management system in this regard.