-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Journal of Microbiology Research
p-ISSN: 2166-5885 e-ISSN: 2166-5931
2014; 4(2): 43-53
doi:10.5923/j.microbiology.20140402.02
A Minireview: Molecular Understanding of HCV Infection Mechanism
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAnimesh Sarker 1, Marufa Nasreen 1, Rafiad Islam 1, Tasnim Ahmed 2, Fahima Rahman 1
1Department of Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University, Santosh, Tangail-1902, Bangladesh
2Department of Biotechnology and Genetic Engineering, East-West University, Dhaka-1200. Bangladesh
Correspondence to: Animesh Sarker , Department of Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University, Santosh, Tangail-1902, Bangladesh.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Hepatitis C (HCV) is a positive polarity single-stranded (ss) RNA virus belongs to Flaviviridae family. It infects about 2% people annually throughout the world (WHO, 2012) and causes both acute and chronic hepatitis consequences permanent liver damage, hepatocellular carcinoma (HCC) and eventually death. The absence of effective means of treatment makes HCV infection a global health hazard. Due to lack of pin point of molecular mechanism, precise drug target and efficient preventive measure is still unclear. Therefore, identifying and understanding mechanistic underpinnings of viral entry, replication, assembly, and budding are crucial in owing to the development of antiviral therapy. Current host-pathogen interactions data and the infection model suggest that RNA dependent RNA polymerase activity of NS5B, along with NS5A and NS3 play central role in HCV infection mechanisms. It has been shown in numerous studies that the interactions between 5′ and 3′ UTRs (Un-translated regions) and the interactions UTRs verses host proteins play fundamental role in regulating replication and translation processes as well as their successive switching.
Keywords: Viral RNA, IRES element, PKR, Signal peptidase, Replicase complex
Cite this paper: Animesh Sarker , Marufa Nasreen , Rafiad Islam , Tasnim Ahmed , Fahima Rahman , A Minireview: Molecular Understanding of HCV Infection Mechanism, Journal of Microbiology Research, Vol. 4 No. 2, 2014, pp. 43-53. doi: 10.5923/j.microbiology.20140402.02.
Article Outline
1. Introduction
- Hepatitis C is one of the most common liver diseases that results from infection with the hepatitis C virus. It is transmitted through blood contact with an infected person. Currently WHO estimated that around 3–4 million people are infected with the hepatitis C virus annually. Approximately 150 million people are chronically infected with hepatitis C virus, and more than 350,000 people die every year from hepatitis C-related liver diseases. HCV infected patients, whether acute or chronic; often have no symptoms [1]. But Persistent infection with HCV often leads to cirrhosis and hepatocellular carcinoma (HCC) [2]. Protective vaccine and other therapeutic alternative are still limited against this virus [3].The HCV is one of the members of Hepacivirus genus and belongs to Flaviviridae family. It‘s a small, enveloped, spherical, positive-sense RNA virus [4, 5]. So far six major genotypes of HCV (HCV 1-6) have been reported yet, each of them contains multiple subtypes. Genotype 3 is mostly prevalent in South-East Asia [6] and genotype 1 is in the US. Genotypes 1 and 4 are less responsive to treatment than other genotypes 2, 3, 5 and 6 [7].The HCV infection solely depends on the interaction between virus and host cell. Further pathogenesis take place due to subsequent host-viral protein-protein (PPI) and protein-RNA (PRI) interactions. As our understanding of molecular interactions between host and the virus increases, more focused HCV infection mechanism being clear. This current review indentifies experimentally validated host-virus interaction partners and sequentially arranged them into a infection model. This conceptual model focuses on recent advances in molecular mechanism of HCV translation, replication as well as their regulatory switch.
2. HCV Lifecycle
- HCV enters the host cell via receptor mediated endocytosis and following pH mediated membrane fusion of the HCV nucleocapsid escapes into the cytoplasm [8]. The positive sense RNA genome is transported to the endoplasmic reticulum (ER), where it is translated to produce viral structural and non-structural proteins. These viral proteins further engage in subsequent rounds of RNA synthesis. While minus (-) strand HCV RNA is synthesized from the parental (+) strand, serves as a template for further replication. These newly generated positive-sense RNA molecules further undergo for second round translation, generating more structural proteins required for RNA packaging and virus assembly [9]. This coordinated process yields virions which becomes maturated in the golgi apparatus and finally secreted via the host secretory pathway [10].
3. HCV Genome and Proteome
- The HCV genome comprises of 9.6 kb and encodes a poly-protein around 3000 aa in length [12]. It carries single open reading frame (ORF) flanked by 5′ and 3′ untranslated regions (UTRs), which are essential for viral translation, replication and infection [13]. The 5′ conserved secondary structure IRES (internal ribosome entry site) directs translation and other 3′ structural elements facilitates newly RNA synthesis [13, 14, 15]. The genomic RNA serves as template mRNA for poly-protein bio-synthesis which is then co and post-translationally processed into four structural protein capsid (C), envelope1 (E1), envelope2 (E2) and (p7) and six non-structural proteins (e.g. NS2, NS3, NS4A, NS4B, NS5A and NS5B) (Figure 2A) [15].5′ UTR: There are four conserved stem-loop structure in 5´ end of HCV RNA which are designated as motif I to IV [16]. Among them motif II and III serve as internal ribosome entry site (IRES), which directs viral translation in a cap-independed manner [17, 18]. This IRES element spans 340 nucleotides (nt) of 5′ UTR and includes a short stretch of the 5′ core coding sequence [19]. Under intracellular condition, motif III forms six distinct stem-loop structures (designated IIIa-f) that provide sites for interaction of numerous host and virus proteins [20]. Most vital motif of IRES element exists in IIId stem-loop structure spanning nucleotides from 253 to 279. This highly conserved IIId region again composed of two short helix separated by a loop E and capped by an apical loop folded into a U-turn motif (Figure 1B) [20, 21].3′ UTR: It is evolutionarily conserved and consists of three principal structured elements spanning around 200 nucleotides from 3′ viral RNA [19, 22]. These elements are highly variable region (VR), a poly U/UC tract and the 3′ X tail (Figure 1C). The third element, 3′ X tail again composed of three stem-loops termed 3′ SLI, 3′ SLII, 3′ SLIII [22]. These elements are conserved and numerous experiments have showed their indispensable association in viral translation and replication [18-22].
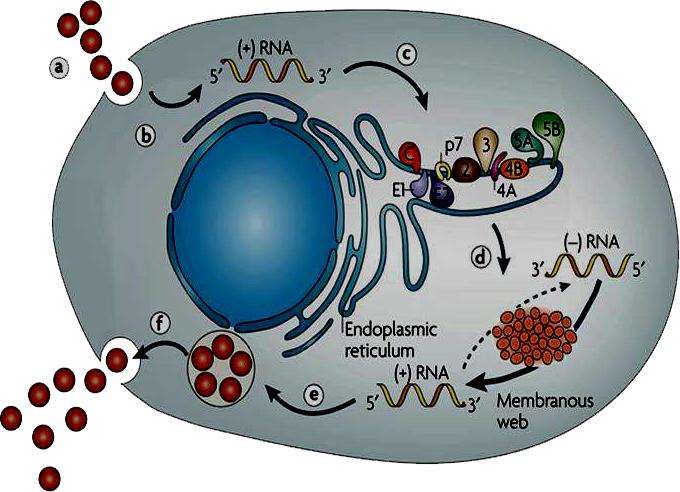 | Figure 1. Overview of molecular mechanism of HCV infection [11]. (a) Virus binding and internalization; (b) cytoplasmic release and uncoating; (c) IRES-mediated translation and polyprotein processing; (d) RNA replication; (e) packaging and assembly; (f) virion maturation and release |
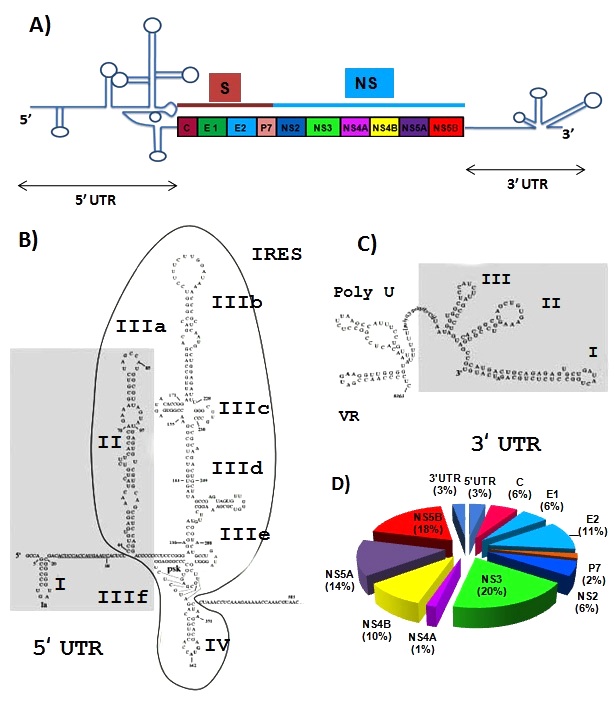 | Figure 2. The Genomic Organization of HCV A) HCV genome composed of four structural protein and six non-structural protein flanked by 5′ and 3′ non-coding sequences; B) Stem-loop structure of HCV 5′ UTR which composed of four conserved motif designated (I to IV); C) Stem-loop structure of HCV 3′ UTR which composed of three conserved element known as highly variable region (VR), a poly U/UC tract and the 3′ X tail and D) Percent Distribution of HCV genomic content. NS5B comprises the highest percentage (18%) and NS4A the lowest percentage (1%) of HCV genome. Around 6% genome of HCV comprises 5′ and 3′ UTR which remain un-translated during translation |
4. Molecular Mechanism of HCV Infection
- Virus Entry: The primary targets of HCV are hepatocyte cells of human and chimpanzee. B cells, dendritic cells and other cell types have also been reported as its alternative target. CD81 a tetraspanin protein, LDL receptor (LDLR), scavenger receptor class B type I (SR-BI) and claudin-1 are most common receptor for HCV endocytosis [36,37].The LDLR is an attractive candidate due to the association of HCV with LDL and VLDL [38]. Together with glycosaminoglycans GAG, the LDLR and other cell surface proteins serve as primary collectors of HCV particles for further targeting to CD81 and additional receptor components (Figure 3). HCV E2 also binds to L-SIGN (liver/lymph node-specific intercellular adhesion molecule-3-grabbing integrin) which is a calcium-dependent lectin expressed on liver sinusoidal endothelial cells that facilitates the infection process by trapping the virus for subsequent interaction with hepatocytes [11]. Another tight junction component claudin-1 (CLDN1) was recently identified as an HCV co-receptor was found to be essential for HCV entry into hepatic cells. CLDN1 acts at the late stage of the entry process, after virus binding and interaction with CD81 [36]. Finally, HCV enters by clathrin-mediated endocytosis [39] with transit through an endosomal, low pH compartment [40] and presumably endosomal membrane fusion [41].
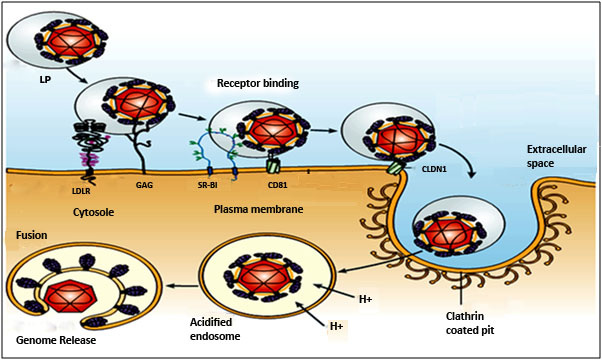 | Figure 3. Hepatitis C virus entry process: HCV particles associate with low- and very-low-density lipoproteins (LP). Low density lipoprotein receptor (LDLR) initially interact virus particle on cell surface. Glycosaminoglycans (GAG), scavenger receptor class B type I (SR-BI), the tetraspanin protein CD81 and claudin-1 (CLDN1) all are involved in the entry of virus particle. Clathrin coated pit mediates endocytosis of virus particle. Acidification of the endosome induces HCV glycoprotein membrane fusion and genome release into the cytosol |
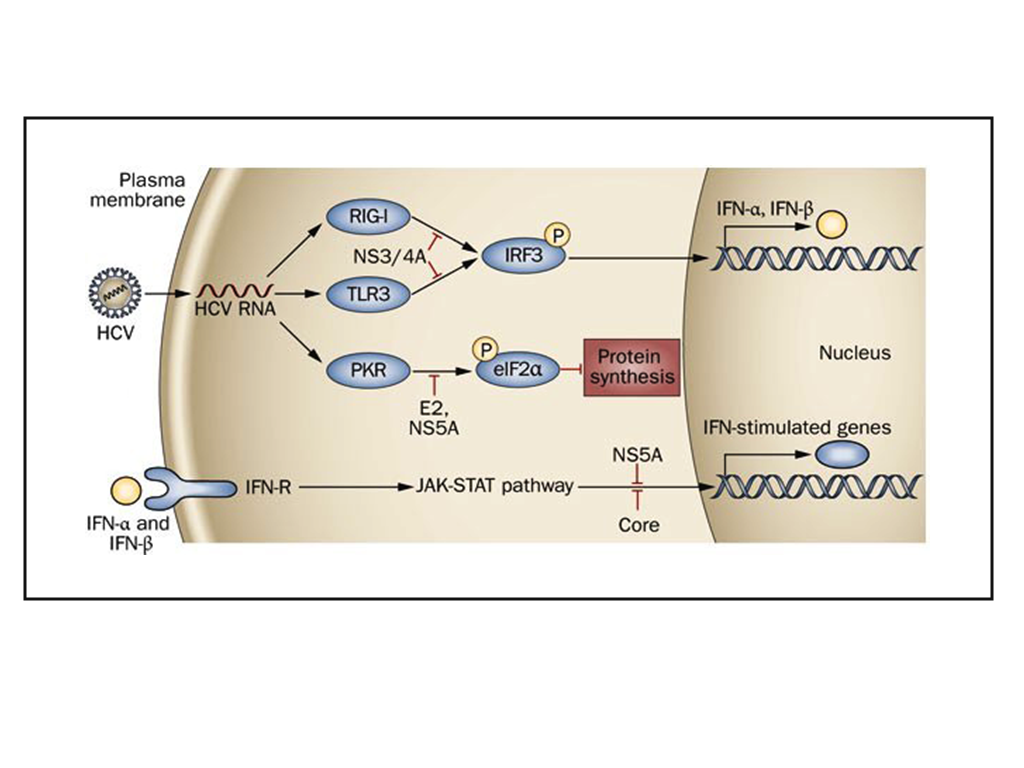 | Figure 4. HCV interferes with IFN signalling, and antiviral effectors. HCV protease NS3/4A blocks downstream signalling of TLR3 and RIG‑I pathogen‑recognition receptors. HCV protein NS5A and Core also block IFN signalling via the JAK‑STAT pathway. E2 and NS5A inhibit PKR, an antiviral effector that leads to inhibition of protein synthesis in infected cells |
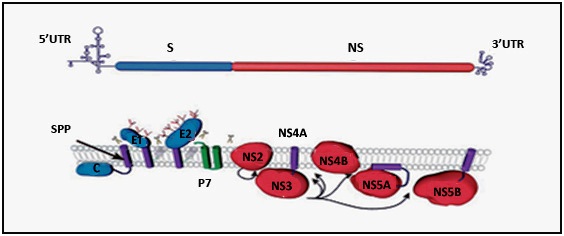 | Figure 5. HCV poly-protein processing and its membrane topology. HCV encodes a single polyprotein with the structural proteins (S) and the non-structural proteins (NS). Scissors indicate cleavages by a host signal peptidase. Arrows indicate NS2-3 and NS3-4A cleavages. The intramembrane arrow indicates cleavage by a host signal peptide peptidase (SPP) |
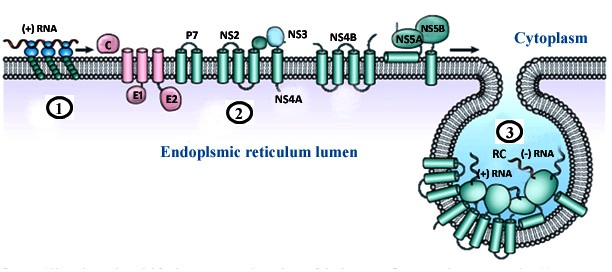 | Figure 6. Topology of the HCV replication complex. (1) The viral genome is translated into a poly-protein (2) Poly-protein processed into structural (pink) and non-structural (green) proteins; (2) (3) viral non-structural protein NS4B induces the formation of membrane alterations which serve as a scaffold for the viral replication complex (RC) assembly |
5. Proposed Mechanistic Model of HCV Infection
- HCV carries only a positive sense RNA during invasion of host cell. It generally enters into the host cell via receptor mediated endocytosis and releases only its genetic material in to the host cell cytoplasm. Primarily, a number of cytoplasmic host proteins unite with viral genome and escort it to endoplasmic reticulum ER. A number of host proteins specially interact with conserved 5′ and 3′ UTR of viral RNA and assists long-range RNA-RNA interaction ensuring its full length genome circularization. Circular genome actually forms partially dsRNA which activates IFNα/β and PKR pathways. dsRNA activated protein kinase R (PKR) phosphorylates translation initiation factor eIF2α and turned off regular protein synthesis. Though, phosphorylated eIF2α could turn back by GADD34-pptase1 de-phosphorylation pathway and revive regular host cell protein synthesis.However, the viral genome is so much tricky and between these phenomenons it can continue its translation through IRES mediated eIF2 independent pathways. In this case, translation initiation factor eIF3 and 40s ribosomal subunit directly bind 5′ IRES and form RNA-40s complex in which initiation codon is placed at the P site of ribosomal complex. Along with other host factor especially eIF5B and 60s ribosome 5′ viral genome form a functional translation initiation complex [46]. Ribosomal complex then carry out complete protein synthesis by eIF5B-GTP and charged tRNA recycling process. Viral genome synthesize as poly-protein which then undergoes for two stride proteolytic cleavage. The first stride structural proteins are processed by host single peptidase and the non-structural proteins are cleaved by viral NS3-NS4 and NS2 proteases.The processed non-structural proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B) are assembled in to replication complex (RC) to synthesize complementary RNA molecule yielding viral genome a double stranded replicative form (RF). The minus strand of RF then utilized by replication complex (RC) to synthesize manifold positive (+) strand RNA. The newly synthesized (+) strand RNA then goes either for second round translation or virion assembly. Structural protein and (+) strand RNA assemble at specialized sites on ER membrane. Further maturation and packaging occurs by transfer of nascent particles across the ER membrane to golgi bodies. Finally, complete packaged virus particle released out by host cell secretary pathway.
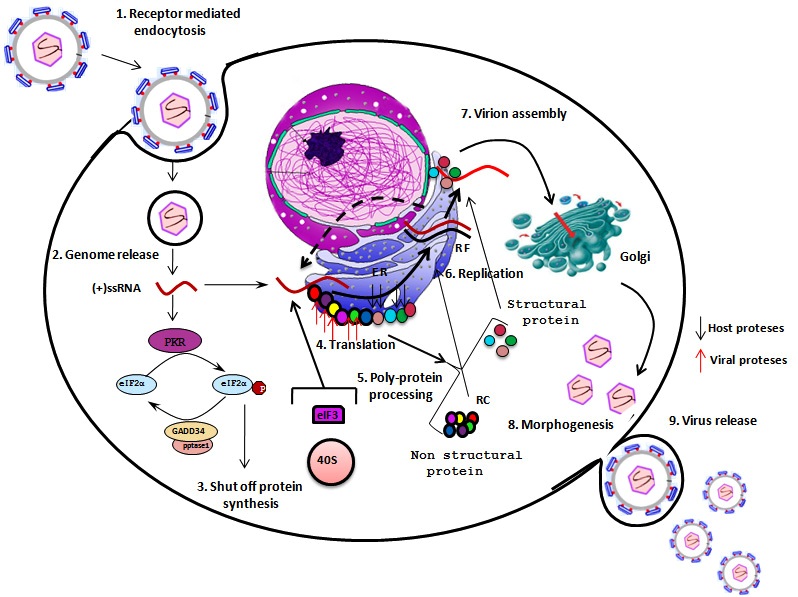 | Figure 7. Hepatitis C virus infection mechanism. 1) HCV enters into host cell via receptor mediated endocytosis. 2) Viral positive strand RNA genome release into in host cell cytoplasm. 3) PKR phosphorylates eIF2 and shut off regular protein synthesis. GADD34-pptase further de phosphorylates eIF2 4) eIF3 and 40s ribosomal subunit directly bind 5′ IRES and along with other factors initiate viral translation in cap independent pathway 5) Viral poly-protein processed by host cell and viral proteses 6) HCV non-structural proteins are assemble into replication complex (RC) that carried out viral replication 7) Structural protein and (+) strand RNA assemble at specialized sites on ER membrane 8) Morphogenesis of complete packaged viral particle 9) Virus particle released out by host cell secretory pathway |
6. Conclusions
- The establishment of HCV infection requires temporal, spatial and mechanistic coordination of the molecular processes of viral replication. Sequences and secondary structures within the HCV genome dynamically interact with viral and host proteins to regulate translation and replication. While we have detailed knowledge of the cis and trans elements required for HCV protein and RNA synthesis, many questions about the coordination of these processes remain to be answered. Continuous development of molecular, biochemical and cell biological tools will facilitate elucidation of the precise mechanisms of first- and second-round translation and minus- and plus-strand replication while advances in proteomics and genomics are likely to provide information about the role of host factors in these processes. The HCV field is at an exciting point where new tools and approaches are advancing our understanding of the intricacies of HCV translation and replication.
References
| [1] | Woods L., Sofosbuvir is much safer drug than interferon, which many patients do not respond to or tolerate. Hepatitis C research and news 2013; 10.1056/NEJMe1303818 |
| [2] | Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet 2012; 379:1245. |
| [3] | Ashfaq U.A., Javed T., Rehman S., Nawaz Z and Riazuddin S., An overview of HCV molecular biology, replication and immune responses. Virology Journal 2011; 161 (8). |
| [4] | Sharma S. D., Hepatitis C virus: Molecular biology & current therapeutic options. Indian J Med Res 2010; 131: p17-34 |
| [5] | MD E. S., Mele A., Mariano A., Risk factors for and incidence of acute hepatitis C after the achievement of blood supply safety in Italy: Results from the national surveillance system. Medical Virology 2012; 85 (3):p433-440 |
| [6] | Ahmadipour M.H., Alavian S. M., Amini S., Azadmanesh K., Hepatitis C Virus Genotypes. Hepatitis Monthly. 2005. 5(3): 77-82. |
| [7] | Simmonds P, Bukh J, Combet C, Deléage G,. "Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes". Hepatology 2005; |
| [8] | Joyce M. A. Tyrrell L. J., The cell biology of hepatitis C virus. Microb Infect 2010; 12 p1-9 |
| [9] | Sklan E. H., Charuworn P., Pang P. S. and Glenn J. S., Mechanisms of HCV survival in the host. Nat. Rev. Gastroenterol. Hepatol 2009; 6, p217–227 |
| [10] | Gastaminza, P. et al. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol 2008; 82: 2120–2129 |
| [11] | Moradpour D., Penin F. and Rice C.M., Replication of hepatitis C virus., NATURE REVIEWS. JUNE 2007. 5:453-463. |
| [12] | Murayama, A., Date, T., Morikawa, K., Akazawa, D., Miyamoto, M., Kaga, M., The NS3 Helicase and NS5B-to-3′X Regions Are Important for Efficient Hepatitis C Virus Strain JFH-1 Replication in Huh7 Cells. J. Virol 2007; 81: 8030–8040 |
| [13] | Friebe, P., and Bartenschlager, R. Lopez C. R. and Herranz A.B. A long-range RNA–RNA interaction between the 5´ and 3´ends of the HCV genome. J. Virol 2002. 76, 5326–5338 |
| [14] | Friebe, P., Lohmann, V., Krieger, N., and Bartenschlager, R. Sequences in the 5′ Nontranslated Region of Hepatitis C Virus Required for RNA Replication., J. Virol 2001.75, 12047–12057 |
| [15] | Lopez C. R. and Herranz A.B. A long-range RNA–RNA interaction between the 5´ and 3´ends of the HCV genome. RNA. 2011 June 27. 15:1740–1752 |
| [16] | Blight, K J., Grakoui, A., Hanson, H. L., Rice C. M.,The Molecular Biology of Hepatitis C Virus. Hepatitis Virus 2002; pp 81-108 |
| [17] | Lukavsky P. J., Structure and function of HCV IRES domains. Virus Res 2009; 139(2-2): 166–171. |
| [18] | Terenin I.M., Dmitriev S.E., Andreev and Shatsky I.N. Eukaryotic translation initiation machinery can operate in a bacterial-like mode without eIF2. Nature Structural & Molecular Biology. August 2008. Volume 15 Number 8: 837-841 |
| [19] | Lopez C. R. and Herranz A.B. A long-range RNA–RNA interaction between the 5´ and 3´ends of the HCV genome. RNA. 2011 June 27. 15:1740–1752 |
| [20] | Jubin R., Vantuno N. E., Kieft J. S., Murray M. G., Hepatitis C Virus Internal Ribosome Entry Site (IRES) Stem Loop IIId Contains a Phylogenetically Conserved GGG Triplet Essential for Translation and IRES Folding. J Virol. 2000; 74(22): 10430–10437 |
| [21] | Barria MI, Gonzalez A, Vera-Otarola J, Leon U, Vollrath V, Marsac D, Monasterio O, Perez-Acle T, Soza A, Lopez-Lastra M. Analysis of natural variants of the hepatitis C virus internal ribosome entry site reveals that primary sequence plays a key role on cap-independent translation. Nucleic Acids Res. 2008. 37: 957–971. |
| [22] | Tuplin A., Struthers M., Simmonds P., Evans D. J., A twist in the tail: SHAPE mapping of long-range interactions and structural rearrangements of RNA elements involved in HCV replication. Nucleic Acids Res. 2012; 40(14): 6908–6921 |
| [23] | Dubuisson J. and Fabiani F.L. Hepatitis C virus proteins. World J Gastroenterol. 2007 May 7.13(17): 2406-2415. |
| [24] | Boulant S., Vanbelle C., Ebel C., Penin F., Lavergne J. P., Hepatitis C Virus Core Protein Is a Dimeric Alpha-Helical Protein Exhibiting Membrane Protein Features. J. Virol. 2005; 79(17): 11353-11365 |
| [25] | Moradpour D., Penin F. and Rice C.M., Replication of hepatitis C virus., NATURE REVIEWS. JUNE 2007. 5:453-463. |
| [26] | Penin F., Dubuisson J., Rey F A., Moradpour D., Pawlotsky J. M., Structural Biology of Hepatitis C Virus. HEPATOLOGY 2004; 39:5–19. |
| [27] | Steinmann E., Penin F., Kallis S., Patel A.H., Bartenschlager R., Pietschmann T. Hepatitis C Virus p7 Protein Is Crucial for Assembly and Release of Infectious Virions. PLoS Pathogens. July 2007; 3 (7): 0962-0971. |
| [28] | Welbourn S., Pause A., The Hepatitis C Virus NS2/3 Protease. Mol. Biol 2007. 9: 63–70. |
| [29] | Zhang C., Zhaohui Cai Z.,Kim YC., Kumar R., Stimulation of Hepatitis C Virus (HCV) Nonstructural Protein 3 (NS3) Helicase Activity by the NS3 Protease Domain and by HCV RNA-Dependent RNA Polymerase. J. Virol 2005; 79 (14): 8687-8697. |
| [30] | Hugle, T., F. Fehrmann, E. Bieck, M. Kohara, H. G. Krausslich, C. M. Rice, H. E. Blum, and D. Moradpour. The hepatitis C virus nonstructural protein 4B is an integral endoplasmic reticulum membrane protein. Virology 2001; 284:70-81. |
| [31] | Elazar M., Liu P., Rice C M., Glenn J. S., An N-Terminal Amphipathic Helix in Hepatitis C Virus (HCV) NS4B Mediates Membrane Association, Correct Localization of Replication Complex Proteins, and HCV RNA Replication |
| [32] | Miller S. and Locker J.K. Modification of intracellular membrane structures for virus replication. Nature Reviews 2008; 6: 363-373 |
| [33] | Brass V., Bieck E., Montserret R., Wölk B., Hellings J. A., Blum H. E., An Amino-terminal Amphipathic α-Helix Mediates Membrane Association of the Hepatitis C Virus Nonstructural Protein 5A. March 8, 2002 The Journal of Biological Chemistry 2002; 277: 8130-8139. |
| [34] | Chinnaswamy S., Murali A., Fujisaki P. L. K and Kao C. Regulation of De Novo-Initiated RNA Synthesis in Hepatitis C Virus RNA-Dependent RNA Polymerase by Intermolecular Interactions. Virology. June 2010. 84(12): 5923–5935. |
| [35] | Bode J G., Brenndo E., Karthe J. and Haussinger D. Interplay between host cell and hepatitis C virus in regulating viral replication. Biol. Chem. October 2009.Vol. 390: pp. 1013–1032. |
| [36] | Evans, M. J. et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007; 446: 801–805 |
| [37] | Bartosch, B. & Cosset, F. L. Cell entry of hepatitis C virus. Virology 2006; 348: 1–12 |
| [38] | D.M. Owen, H. Huang, J. Ye, M. Gale Jr., Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipo-protein receptor. Virology (2009). |
| [39] | Blanchard, E. et al. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 2006; 80: 6964–6972. |
| [40] | Tscherne, D. M. et al. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 2006; 80: 1734–1741. |
| [41] | Gibbons, D. L. et al. Conformational change and protein–protein interactions of the fusion protein of Semliki Forest virus. Nature 2004; 427:320–325 |
| [42] | Sklan E.H., Charuworn P., Pang P.S. and Glenn J.S. Mechanisms of HCV survival in the host. Nature reviews 2009. volume 6: 217-227. |
| [43] | Schneider R. J. and Mohr I., Translation initiation and viral tricks. TRENDS in Biochemical Sciences. March 2003., Vol.28 No.3: p130-136. |
| [44] | Robert, F. et al. Initiation of protein synthesis by hepatitis C virus is refractory to. |
| [45] | Reduced eIF2 GTP Met-tRNAiMet ternary complex availability. Mol. Biol. Cell 2006 ;17:4632–4644. |
| [46] | Terenin I.M., Dmitriev S.E., Andreev and Shatsky I.N. Eukaryotic translation initiation machinery can operate in a bacterial-like mode without eIF2. Nature Structural & Molecular Biology. August 2008. Volume 15 Number 8: 837-841. |
| [47] | Taylor, D. r. et al. inhibition of the interferon‑inducible protein kinase PKR by HCv E2 protein. Science 1999; 285: 107. |
| [48] | Noguchi, T. et al. effects of mutation in hepatitis C virus nonstructural protein 5A on interferon resistance mediated by inhibition of PKR kinase activity in mammalian cells. Microbiol Immunol 2001; 45: 829–840. |
| [49] | M. Gale Jr., E.M. Foy, Evasion of intracellular host defence by hepatitis C virus. Nature 2005; 436: 939-945. |
| [50] | Ke PY. and Chen S .S.L. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. The Journal of Clinical Investigation. January 2011.Volume 121 Number 1: 37-56. |
| [51] | Harris E. and Pena J. Dengue Virus Modulates the Unfolded Protein Response in a Time-dependent Manner. The Journal Of Biological Chemistry. January 19, 2011.286(16): pp. 14226–14236. |
| [52] | Andreev, D.E., Terenin, I.M., Dunaevsky, Y.E., Dmitriev, S.E. & Shatsky, I.N. A leaderless mRNA can bind to mammalian 80S ribosomes and direct polypeptide synthesis in the absence of translation initiation factors. Mol. Cell. Biol. 26, 3164–3169 (2006). |
| [53] | Terenin I.M., Dmitriev S.E., Andreev and Shatsky I.N. Eukaryotic translation initiation machinery can operate in a bacterial-like mode without eIF2. Nature Structural & Molecular Biology. August 2008. Volume 15 Number 8: 837-841. |
| [54] | Fraser C.S. and Doudna J.A. Structural and mechanistic insights into hepatitis C viral translation initiation. Nature Reviews. January 2007. Volume 5: 29-38. |
| [55] | Hellen C.U.T., IRES-induced conformational changes in the ribosome and the mechanism of translation initiation by internal ribosomal entry., Biochim Biophys Acta. 2009. 1789(9-10): 558–570. |
| [56] | Tischendorf JJW. 2004. Polypyrimidine tract-binding protein (PTB) inhibits Hepatitis C virus internal ribosome entry site (HCV IRES)-mediated translation, but does not affect HCV replication. Arch Virol 149:1955-1970. |
| [57] | Hahm B, Kim YK, Kim JH, Kim TY, Jang SK. 1998b. Heterogeneous nuclear ribonucleoprotein L interacts with the 3' border of the internal ribosomal entry site of hepatitis C virus. J Virol 72:8782-8788. |
| [58] | Song Y., Bindereif A. and Niepmann M. Regulation of Hepatitis C Virus translation by the viral internal ribosome entry site and the 3′-untranslated region. Jan 2006. |
| [59] | Isken O., Baroth M., Grassmann C.W.Weinlich S., Ostareck D.H., Antje Ostareck-Lederer A.O. And Behrens SE. Nuclear factors are involved in hepatitis C virus RNA replication. RNA. 2007.13(10):1675-1692. |
| [60] | Honda M. Hepatitis C Virus Translation And Cell Cycle. Biomedical Reviews. 2004. 15: 37-46. |
| [61] | Pacheco A. and Salas E.M. Insights into the Biology of IRES Elements through Riboproteomic Approaches. Journal of Biomedicine and Biotechnology.2009. Article ID 458927 |
| [62] | Yuan Z., Lan S., Wang H., Jiang H., Mao H., Liu X and Zhang X. Direct interaction between α-actinin and hepatitis C virus NS5B. FEBS Letters 2003. 554: 289-294. |
| [63] | Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology 2004; 39: 5-19. |
| [64] | Morikawa K., Lange C. M., Gouttenoire J., Meylan E., and Penin F., Nonstructural protein 3-4A: the Swiss army knife of hepatitis C virus. Journal of Viral Hepatitis. 2011. 18: 305–315. |
| [65] | Dubussion J. Hepatitis C virus proteins. Gastroenterology 2007: 13(17): 2406-2415. |
| [66] | Schwartz, M. et al. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell 9, 505–514 (2002). |
| [67] | Antonny, B. Membrane deformation by protein coats. Curr. Opin. Cell Biol. 2006; 18, 386–394 . |
| [68] | Lundin, M. Topology and Membrane Rearrangements of the Hepatitis C Virus Protein NS4B (Larseries Digital Print AB, Stockholm, 2006); 1–55 |
| [69] | McMahon, H. T. & Gallop, J. L. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 2005. 438; 590–596. |
| [70] | Zhong, W., Uss, A. S., Ferrari, E., Lau, J. Y. N. & Hong, Z. De novo initiation of RNA synthesis by hepatitis C virus nonstructural protein 5B polymerase. Journal of Virology 2000; 74, 2017-2022 |
| [71] | Luo, G., Hamatake, R. K., Mathis, D. M., Racela, J., Rigat, K. L., Lemm, J. & Colonno, R. J. (2000). De novo initiation of RNA synthesis by the RNA-dependent RNA polymerase (NS5B) of hepatitis C virus. Journal of Virology 2000; 74, 851-863. |
| [72] | Bartenschlager R. and Lohmann V, Replication of hepatitis C virus., Journal of General Virology 2000; 81: 1631–1648. |
| [73] | Ndjomou J., Park I., Ying Liu Y., Mayo L.D., Johnny J. and He J.J. Up-Regulation of Hepatitis C Virus Replication and Production by Inhibition of MEK/ERK Signaling. PLoS ONE. 2009; Volume 4, Issue 10: e7498 |
| [74] | Jones D. M. And Mclauchlan J. Hepatitis C Virus: Assembly and Release of Virus Particles. Journal Of Biological Chemistry. 2010; 285: 22733-22739 |
| [75] | André, P., Perlemuter, G., Budkowska, A., Bréchot, C. & Lotteau, V. Hepatitis C virus particles and lipoprotein metabolism. Semin. Liver Dis 2005; 25: 93–104 |