-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Basic Sciences of Medicine
p-ISSN: 2167-7344 e-ISSN: 2167-7352
2015; 4(1): 5-11
doi:10.5923/j.medicine.20150401.02
An Induced Disequilibrium between the AT1 and B2R Impairs the Trophoblastic Motile Capacity: A Cellular Insight of Preeclampsia
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAlberto Leguina-Ruzzi
Faculty of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile
Correspondence to: Alberto Leguina-Ruzzi , Faculty of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Preeclampsia affects 2-3% of the pregnancies worldwide and is one of the main causes of maternal death and premature birth. Its first stage is related to the invasion of the maternal decidua and the remodeling of the uterine spiral arteries by the extravillous trophoblast. An enhancement of the systemic and local renin-angiotensin system has been identified in PE patients, and the RAS counter-regulatory kallikrein–kinin system is expressed in extravillous trophoblasts. The aim of the present study was to evaluate the effect of a misbalance provoked by exogenous Angiotensin II (AII) and blockade of the B2R receptor by the non-peptide antagonist Bradyzide (BDZ) on the migratory phenotype and and on invasive capacity of HRT8/SVneo cells, a well accepted trophoblast in vitro model. HTR-8/SVneo cells express Kalicrein, Kininogen, AT1, AT2, B1R and B2R. AII, AII+BDZ and losartan induced marked changes in the length and shape of filopodias. AII and AII+BDZ reduced significantly the number of filopodias, the migration and invasion capacity. Losartan reverted the effect of AII on the number of filopodias and the invasion index. In conclusion an induced disequilibrium between the AT1 and B2R alters morphological and motile characteristics that could explain in part the cellular aspects of the impaired trophoblastic invasion present on preeclampsia and could be a potential treatment target for preeclampsia.
Keywords: Preeclampsia, AT1, B2R, Trophoblast, RAS
Cite this paper: Alberto Leguina-Ruzzi , An Induced Disequilibrium between the AT1 and B2R Impairs the Trophoblastic Motile Capacity: A Cellular Insight of Preeclampsia, Basic Sciences of Medicine , Vol. 4 No. 1, 2015, pp. 5-11. doi: 10.5923/j.medicine.20150401.02.
Article Outline
1. Introduction
- Preeclampsia (PE) is a syndrome that affects between a 2 to 3% of the pregnancies worldwide and is one of the major causes of maternal death and premature birth [1]. The main feature of PE is “de novo” hypertension accompanied by proteinuria. There are several evidences that show a genetic predisposition to PE, but the interaction with the environment is crucial for the expression and progression of the syndrome [2]. The current gold standard treatment is the termination of pregnancy, option that is not always the most suitable alternative [3], for that reason it is important to understand the molecular processes that underlay the PE as potential treatment targets.Even when the physiopathology of PE is yet not fully understood the two stages model is highly accepted. In an early silent phase there is a shallow remodelation of spiral arteries, which has been attributed to an unfavorable immunological reaction, a deficiency of adhesion molecules, of local vasodilatation, underlying maternal cardiovascular, metabolic, coagulation factors, among others. The ischemic placenta sheds to the maternal circulation syncytial microparticles, reactive oxygen species, antiangiogenic and vasoconstrictor factors pro inflammatory cytokines leading to clinical overt second stage of the disease [4].Considering that the first stage of PE as a cellular process related to the invasion of the maternal decidua and the uterine spiral arteries by the extravillous trophoblast (EVT) we were interested in identifying new factors that contribute to impair EVT invasion. This cellular phenomenon has been associated to several pathologies, as intrauterine growth retardation, preterm birth, placenta accreta and the preeclampsia syndrome [5-9].A disequilibrium of the systemic and local renin-angiotensin system has been characterized in PE patients. There is a high production of Angiotensin II (AII) and activity of AT1 receptor in chorionic villi [10]. There are strong evidences that present this vasoconstrictor as an inductor of apoptosis and as an inflammatory agent in several tissues, among them the feto-maternal-placental interface [11, 12]. Moreover there is clear evidence of the presence of the AT1 and AT2 receptors in the feto-maternal interphase; since these receptors have opposite signalling pathways, the effect of AII on the trophoblast could have yet undescribed effects [13].In spite of all we know about the function of the AII, there is only one study that shows its capacity to reduce trophoblast invasion through the AT1R [14]. This is a relevant issue as preeclampsia is associated with the generation of agonistic AT1R autoantibodies, which could exert vasoactive and other unknown actions [15].On the other hand the expression of the local kallikrein –kinin system, a counteregulatory system to the RAS, has been documented in the chorionic villi, and bradykinin exerts a vasodilator effect on fetal vessels [16]. In HTR8/SVneo bradykinin stimulates the migratory and invasive capacity of cells by acting on the B2R receptor [17].The aim of the present study was to evaluate the effect of a misbalance of the RAS and the kallikrein-kinin systems by incubating HRT-8/SVneo cells with exogenous AII in combination with a B2R antagonist (Bradyzide [BDZ]), on the migratory and invasive capacity of HTR-8/SVneo cells. These cells are immortalized line of first trimester extravillous trophoblast, and constitute a valid cellular model. Our results permitted us to characterize the cell line in terms of the expression of the vasoactive receptors and of the cytotoxicity of our stimuli. Through the use of an AII AT1R receptor blocker we confirmed that the effect of the peptide is exerted via the AT1R. We demonstrated that the combination of AII and BDZ reduces migration and invasion. Moreover, our stimuli were able to disturb the number and shape of filopodias.A study that tested this intervention, in an animal model, shown an impaired gestational outcome without obtaining a full PE phenotype [18]. However our data contributes to underscore the pivotal role of a normal or defective equilibrium of the RAS/KKS systems in determining an adequate or a defective placentation process.
2. Materials and Methods
- We followed the protocols previously described when studying the effect of bradykinin on migration and invasion [17].HTR-8/SVneo cell cultureHTR-8/SVneo trophoblast cells were kindly donated by C.H. Graham, Queen's University, Kingston, Canada. The cells were obtained from first trimester human placenta (8-10 wk gestation) and immortalized by transfection with a cDNA construct that encodes the simian virus 40 large T antigens. Though non tumorigenic and non metastatic, the cells are highly invasive in vitro and exhibit phenotypic properties of extravillous cytotrophoblasts, including the expression of cytokeratins 7, 8, and 18, placental alkaline phosphatase, urokinase-type PAR, human leukocyte antigen (HLA) framework antigen W6/32, IGF-II mRNA and protein, and the integrin profile characteristic of invasive cytotrophoblasts. When plated on Matrigel these cells express HLA-G. The AT1R has been also demonstrated. A previous study of this laboratory identified in them kallikrein, its two receptors (B1R and B2R), as well as their capacity to incorporate kininogen from the culture medium. Cells were cultured as described. Briefly, cells were grown in RPMI 1640 (Sigma-Aldrich) supplemented with 10% FBS (Gibco) and 50 μg/ml gentamicin (Gibco) in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Cells at passage 6-25 were used to perform all the experiments.ImmunocytochemistryImmunocytochemistry was performed for AT1R, AT2R, B2R, kallikrein, kininogen, cytokeratin and Ki67. The dilution and origin of the antibodies were: anti-AT1 (1:200, SantaCruz), anti-AT2 (1:200, Santa Cruz), anti-B2R mouse monoclonal (1:4,000, BD Transduction Laboratories), anti-pancytokeratin mouse monoclonal, (1:100, Sigma-Aldrich), anti-Kal (generated at the laboratory) anti-KK (1:500, Boehringer) and anti-Ki67 mouse monoclonal (1:1000, Cell Signaling).Cells were seeded at a density of 80,000 cells/well on cover slides in 24-well plates (Nucleon Surface, Nunc), in RPMI 1640, supplemented with 1% FBS, 50 µg/ml gentamicin in a humidified atmosphere of 95% air and 5% CO2 at 37°C. After 18 hours, cells were rinsed 3 times with cold PBS and fixed with methanol at -20°C for 20 minutes, equilibrated 3 times with 50 mMol/L TRIS-HCl PBS buffer pH 7.8 and incubated with 10% H2O2 for 5 minutes to block endogenous peroxidases. Cells were incubated with protein block (Cas-Block, Zymed Laboratories) for 30 minutes in a humidified chamber, and incubated with the respective antibodies at the aforementioned concentrations for 18 hours at 4°C. Cells were immunostained with the biotin-streptavidin peroxidase system (Dako LSAB+System-HRP), incubated with 0.1% of 3-3'-diaminobenzidine (Sigma-Aldrich) in the presence of 0.05% H2O2 for 15 minutes, stained with Harris Haematoxylin (Sigma-Aldrich) and finally mounted with glycerin-gelatin (Kaisers, Merck).Western blot analysisTotal proteins from HTR-8/SVneo cells were extracted using 20 mM Tris-HCl buffer containing 10 mM EDTA, 2 mM phenylmethylsulfonylfluoride, 5 µM leupeptin, 50 μg/ml soybean trypsin inhibitor and 0.02% NaN3 at pH 7.4. Lysed cells were left for 20 minutes on ice, centrifuged at 14,000 rpm for 20 minutes at 4°C and finally stored at -70°C. Protein content was determined according to the Lowry method.Western blots for AT2 were performed as previously described. Equal amounts of protein (50 μg/lane) were separated using 10% SDS-PAGE under reducing conditions and transferred to nitrocellulose membranes (Biorad, Hercules, CA), blocked with 5% nonfat dry milk in PBS-0.1% Tween-20 buffer (PBS-T) and incubated overnight at 4°C with the same primary antibody AT2 mouse monoclonal (1:1000, Santa Cruz, USA) diluted in blocking buffer. The membranes were washed six times for five minutes in PBS-T buffer, incubated with HRP-conjugated anti-mouse secondary antibodies (1:3000, Biorad, Hercules, CA) for one hour at room temperature and developed with chemiluminescence reagent (NEL-103, Western Lighting, Perkin-Elmer, MA). Membranes were exposed to CLxPosure film (Pierce, Rockford, IL). Equal protein loading was confirmed with Ponceau-S red staining (Sigma, St. Louis, MO). Images were scanned at 16-bit/600 dpi resolution with an Epson Perfection 3490 scanner (Epson Corporation, CA), saved as tiff files and calibrated to an optical density scale.Proliferation and viability assaysCells were seeded at a density of 80,000 cells/well on cover slides in 24-well plates and incubated for 18 hours in medium 1% FBS with and without the Angiotensin II and Bradyzide treatments. Immunocytochemistry was performed as previously described using antibody to Ki67, a nuclear protein expressed in the active phases of the cell cycle. The proliferation index was represented by the percentage of cells with positive staining within total cells; the proliferation ratio represented the proliferation index of the treated/untreated cells in each experiment.In addition, cell viability was evaluated using the MTS assay (Promega). Cells were placed in complete medium at a concentration of 15,000 cells/well in 96-well plates for 24 hours. To determine the effect of treatments at 18 hours cells were incubated in reduced serum with 1% FBS; the release of formazan was read after 2 hours of incubation with the MTS substrate at 495 nm by spectrophotometry (Multiskan EX, LabSystem). All experiments were performed in triplicate and repeated at least three times.Migration assayCell migration was studied using the wound-healing assay under the effect of Angiotensin II with and without the B2R antagonist. HTR-8/SVneo cells (100,000) were seeded in each well on cover slides in 24-well plates containing 500 μl complete medium for 24 hours. Cells were then washed 3 times with PBS and the wound was generated by removing cells in the centre of the well with a sterile pipette tip; the unattached cells were washed away with PBS. The experiments were performed in reduced serum (1% FBS). Cells were incubated for 18 hours under the different treatments. Cell migration was studied under the effect of 1x10-8M, 1x10-7M, 1x10-6 of Angiotensin II. In addition cells were preincubated for 30 minutes with Losartan 50µM (Sigma) and AII combined with Bradyzide 1x10-7M (Sigma) and then incubated with their respective ligand for 18 hours in all assays.The migration assay was performed 3 times in triplicate. After fixing the cells with PFA 4% and staining them with Crystal Violet at 1%, three images were obtained along the wound, with a Nikon TMS inverted microscope connected to a Nikon CoolPix 4500 camera; the distance between the borders of the wound were quantified using ImageJ v.1.34 software. The migration index was defined by the distance between borders in response to each study condition divided by the distance observed in response to medium alone.Invasion assayBriefly, transwell inserts containing membranes with 8 μm pore size (Nunc) were coated with matrigel (BD Biosciences) as per the manufacturer's instructions. Cells (30,000) were spread over the matrigel (1:10) in 200 µl of medium with reduced serum (1% FBS), and 800 µl of culture medium was added to the lower chamber. Cell invasion was studied under the effect of Angiotensin II, of AII plus a B2R non-peptide antagonist Bradyzide (BDZ, Sigma)-and finally with the AT1 antagonist Losartan (Sigma). The different agents were added on the upper and lower chambers of the invasion well.For assessing the number of invaded cells, the filters were stained with anti-cytokeratin antibody and haematoxylin and mounted on cover slides with Kaiser's, glycerin-gelatin (Merck). Each experiment was performed in duplicate and was repeated 3 times. The inserts were examined by a blinded observer with an AX.10 microscope (Carl Zeiss) attached to a Nikon CoolPix 4500 camera; 20 photographs, were obtained per insert, and the number of cells in the underside of the filter was counted. Results are expressed as invasion index, where the level of invasion was defined as cells invading in response to BK divided by cells invading in response to medium alone.Fluorescence StainingThe identification of morphological changes in the actin cytoskeleton was performed using FITC-conjugated phalloidin (Molecular Probes). Briefly, 10,000 cells were seeded on coverslips, and incubated with culture medium with AII, BZD, Losartan and control conditions for 18 hours. The cells were rinsed with PBS and fixed in 4% paraformaldehyde in PBS for 15 minutes at room temperature. To quench the excess of aldehyde, 0.1 M glycine in PBS was added for 5 minutes. Cells were permeabilized with 0.1% Triton-X100 in PBS for 1 min and incubated with FITC-labelled phalloidin (1:100) in PBS for 15 minutes. Finally cells were rinsed 3 times for 5 minutes in PBS, then incubated with DAPI (ThermoScientific) 1:1000 for 10 minutes, rinsed 3 mores times and mounted for microscopy with Fluoromont G. As above, microphotographs were obtained with an AX.10 microscope attached to a Nikon CoolPix 4500 camera adapted with an epi-ilumination mercury lamp. A blinded observer counted the number of filopodia connecting two cells in one 130-µm2 circle per cover slip in at least 20 images from each experiment. The total number was then determined with ImageJ software, version 134. Statistical AnalysisResults are expressed as mean ± SE. One-way analysis of variance, followed by Tukey's Multiple Comparison post-hoc test, was used to test for differences between the different interventions and study periods with Graphpad (Prism Inc.). A p < 0.05 was considered significant.
3. Results
- Immunocytochemical detection of AT1 and B2R in HTR-8/SVneo cellsAll cells expressed the Angiotensin II receptors 1 and 2 (AT1R and AT2R) the bradykinin receptor 1 and 2 (B1R and B2R), Kallikrein (Kal) and low molecular weight Kininogen; the pattern for all of them was diffuse in the cytoplasm and punctuate in the membrane (Figure 1A). The immunohistochemistry was performed in 1% BFS since previous work showed that under this condition cells expressed kininogen trapped from the culture medium; in order to conserve the autocrine capacity of kallikrein to generate bradykinin, this was used in the experiments described below [17]. We confirmed the expression of AT2 through Western Blot (Figure 1B).
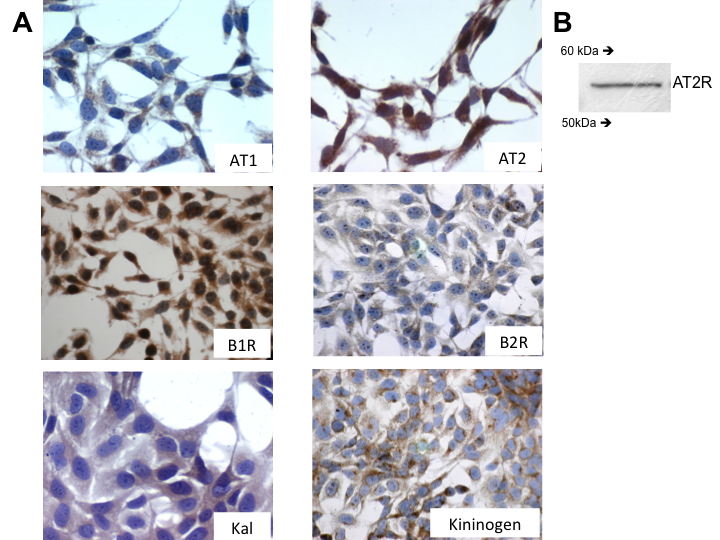 | Figure 1. Inmunoreactivity for AT1, AT2, B1R, B2R, Kal, and KK antibodies in HTR-8/SVneo cells: A. Expression of AT1, At2, B1R, B2R, Kal and Kininogen in cell with 1% BFS and zoom 40X. B. WB that confirms the expression of AT2 molecular weight 54kDa |
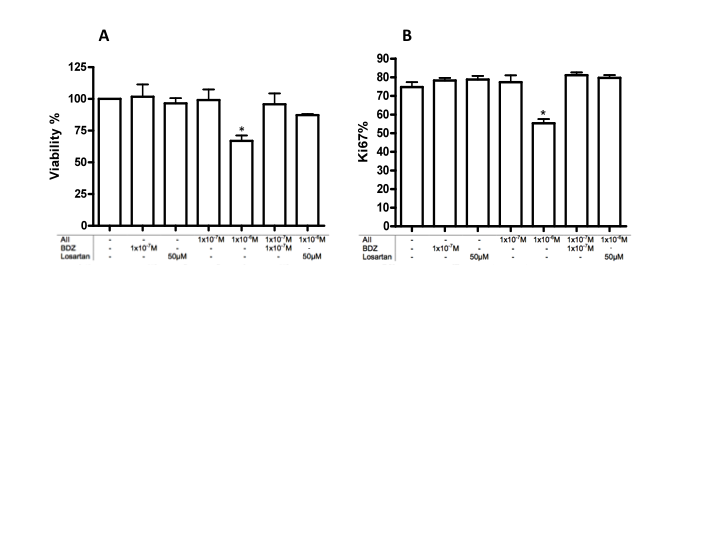 | Figure 2. Assessment of cell viability and proliferation under Angiotensin II and Bradyzide stimulus. A. MTS viability assay for 10,000 cells stimulated for 18 hours. B. Proliferation Ki67 assay quantification that confirms the effect of AII and BDZ in the cell survivor. n=3 *p<0.05 |
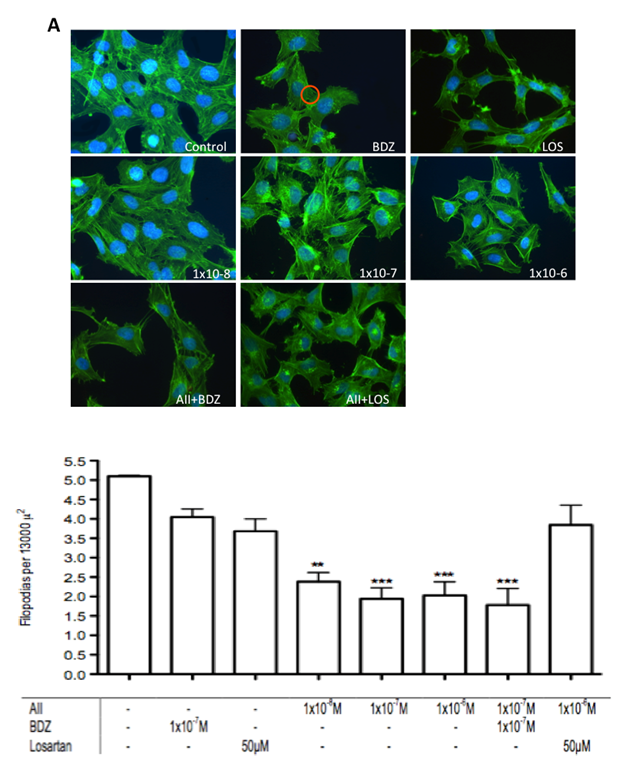 | Figure 3. Effect of the misbalance of RAS and the KKS on filopodias number. A. Reduction of the actin cytoplasmic scaffold, shorter, nubby and/or flaccid filopodias. FITC fluorescente staining and images taked at 40X. B. The red circle depicts the area of 13000  within which the filopodias were counted. n=3 **p<0.001, ***p<0.0001 within which the filopodias were counted. n=3 **p<0.001, ***p<0.0001 |
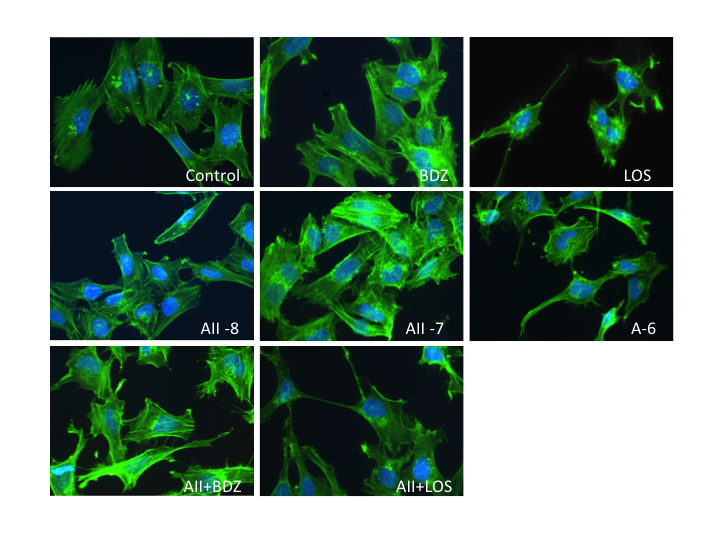 | Figure 4. Effect of the misbalance of RAS and the KKS in cell morphology. Changes in the size and actin structure before 18 hours of stimulation with AII and BDZ. Zoom 40X |
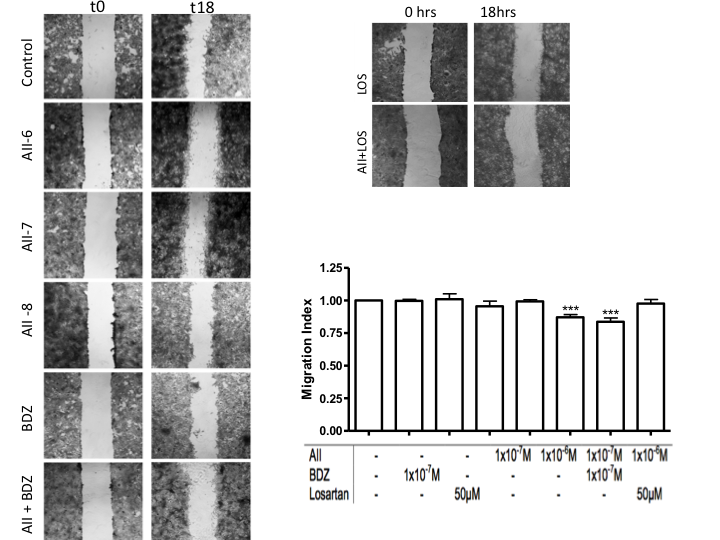 | Figure 5. Effects of the misbalance of RAS and the KKS on the migratory capacity of HTR-8/SVneo cells. Representative images in zoon 20X obtained along the wound, at 0 and 18 hours of stimulation. Quantification of the Would Healing in the graphic. n=3 ***p<0.0001 |
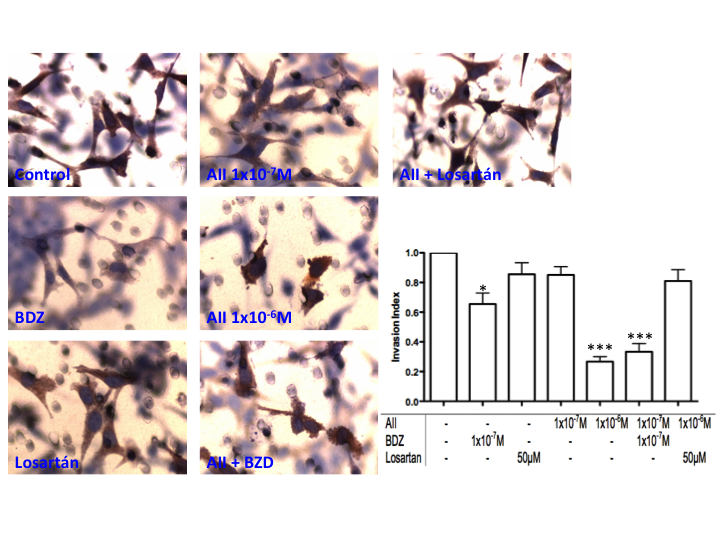 | Figure 6. Effects of the misbalance of RAS and the KKS on the invasive capacity of HTR-8/SVneo cells. Representative images at 20X zoon of invaded inserts showing the effect of AII and BDZ after 18 hours of stimulation. We evaluated by the number of cells focused on the underside of the filter. Blurry cells correspond to non-invading cells in the upper surface of the filter. n=3 *p<0.05, ***p<0.0001 |
4. Discussion
- The present study provides evidence of the role of Angiotensin II as an anti migratory and anti invasive peptide that is potentiated by a RAS/KKS misbalances in HTR-8/SVneo trophoblasts. By using a specific non peptidic blocker, we demonstrated that the effect of AII is exerted via AT1 receptor, evidence that has a clinical relevance in terms of new potential combined treatments for PE.Even when the AII inhibitory effect in terms of invasion, through the PAI-1 synthesis, has been previously reported in HTR-8/SVneo [14] our findings emulate the physiological state, in which the unbalanced RAS/KKS acts as an enhancer system. More importantly, considering our results and the evidence showing that in PE there is an attenuated vasodilatory response to bradikynin [19], further studies in PE animal models could evaluate the effect of the combined treatment; inhibiting AT1 and activating B2R.At the same time that the physiology of these cells was affected by the stimulus, we showed that the cellular morphology was drastically altered and the number of filopodias reduced. Moreover, Losartan, an antihypertensive drug that was used to dissect the AII via of action, changed significantly the actin structure. Currently the preeclampsia management is the pregnancy termination [3] supported by the control of blood pressure and coagulation administrating aspirin and methyldopa [20] however, this strategy does not improve significantly the clinical outcome and their use is associated with several side effects [21]. Even when the management of RAS alterations appears to be a logical choice, given the strong evidence about an excessive AT1 receptor activation during preeclampsia and the studies carried on animal models [22-24], there is a complete lack of clinical studies testing low doses of Losartan in preeclampsia patients. Considering the ethical implications and the high risk that could carry a trial of this type, our in vitro study provides support for the design of a highly controlled clinical evaluation.A pending question is whether this effect is only attributable to Losartan, or is shared by other AT1R receptor blockers however, considering the widely use of Losartan and its specificity, this drug is a good toll for the study of AT1 signaling.Even when the HTR-8/SVneo line is extensively characterized we have been the first to show that they express the AII receptor 2, sustaining the relevance of studying the trophoblast physiology using this model. Under that context it is highly accepted the role of AT2 receptor as a counter regulatory protective component in the blood pressure and sodium excretion control [25]. The expression of this receptor is lower than its counterpart AT1 [26], however the evidence of its microvascular nitric oxide dependent dilator action has gained importance for the investigation of future therapeutic applications in the hypertension field [7]. Even when the field advances have provided important evidence of AT2 physiology, no studies have evaluated the role of this receptor in the motile capacities of vascular cell; the closer approximation to this matter has been related to cancer, in which AT2 signaling attenuates the growth and migration of murine pancreatic carcinoma [28].Further studies are required to evaluate the physiological effect of the selective activation or over expression of AT2 in HTR-8/SVnew, as a potential counter response to the AT1 signaling in vascular migration, particularly of the trophoblast. An unexpected observation of this study is the reverse autoregulation of the AT1R and B2R, which induces a “vicious cycle” that potentiates the AT1 signaling.Though our results were obtained in vitro in an immortalized trophoblast cell line we demonstrated that a disequilibrium between the AT1 and the B2R alters a serie of physiological features related to migration and invasion, in vivo this situation could derive from AT1R agonistic autoantibodies and to heterodimerization of AT1R and B2R [15], or from excessive generation of Angiotensin II caused by the angiotensinogen M235T polymorphism [29] or through the over oxidized angiotensinogen form that modulates angiotensin release [30].
5. Conclusions
- The evidence given in this work shows that an induced disequilibrium between the receptors AT1 and B2R, using exogenous agonist and inhibitor, alters the trophoblast morphological and motile characteristics that could explain in part the cellular aspects of the impaired trophoblastic invasion present in Preeclampsia. Furthermore the clinical relevance of modulate AT1 activity under a pathophysiological RAS/KKS misbalance represents a novel approach for the early treatment of PE development that could benefit the mother and the fetus development.
ACKNOWLEDGEMENTS
- The author is grateful to CH Graham from Queen's University, Kingston, Ontario, Canada for his gift of HTR-8/SVneo cells to Dr. Gloria Valdés who supervised during the second semester of 2011 the doctoral research unit of which this work was in part made, to MP Ocaranza PhD for her donation of Losartan, C Vio MD for the AT2 antibody, P Smith to help us with the Ki67 assay and to Jenny Corthorn (deceased). The research was supported during the second semester of 2011 by internal funds of the Laboratorio de Desarrollo Placentario, Centro de Investigaciones Médicas, PUC.