-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Basic Sciences of Medicine
2012; 1(3): 6-11
doi: 10.5923/j.medicine.20120103.01
A Molecular Genetic Study of Autosomal Recessive Primary Microcephaly in three Pakistani Kindred
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLRabiya Nawab , Saba Irshad
Institute of Biochemistry and Biotechnology University of the Punjab Lahore, 54590, Pakistan
Correspondence to: Saba Irshad , Institute of Biochemistry and Biotechnology University of the Punjab Lahore, 54590, Pakistan.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Microcephaly (MCPH) is a condition with architecturally normal brain but reduced head of occipito-frontal circumference below -2 or -3 standard deviation. Primary microcephaly is present at birth leading to non-progressive mental retardation, affecting 2-2.5% of the total population and is more prevalent in Asia and Arab than Europe. About 50% individuals with primary microcephaly have an ASPM mutation that is the most common cause of MCPH in Pakistan. In this present study exclusion mapping of three different families of primary microcephaly was done. DNA was isolated from three affected families (MCP1, MCP2 and MCP3) and PCR was followed by non-denaturing polyacrylamide gel electrophoresis. One family MCP1 showed complete linkage with locus 5 (MCPH5) on chromosome 1q31. Second family MCP2 linked at locus 2 on chromosome 19q13.1-13.2 and third family MCP3 did not linked with any of the known locus (MCPH1-MCPH7). The heterozygous pattern of affected individuals was found for MCP3 family during screening of all known loci. This finding suggested the plausibility of a novel locus, so genome wide search of MCP1 family should be supportive in future, which would lead to the mapping of a novel region associated with primary microcephaly.
Keywords: Primary Microcephaly, MCPH5, MCPH2, Homozygosity
Article Outline
1. Introduction
- Microcephaly (MCPH) is a finding of reduced head due to less neuron but architecturally normal brain where cerebral cortex shows the greatest size reduction. After the age of six months, condition leads from mild to severe cognitive impairment, without major motor delay; mild seizures; narrow sloping forehead with reduced cranial size; absence of malformations in other organ systems; and normal growth[1]. Primary microcephaly is a neurodevelopment disorder characterized by two principal features, congenital microcephaly that is present at birth and non-progressive mental retardation. MCPH affects 2-2.5% of the total population[2]. Recently the birth prevalence of primary microcephaly is calculated 1/10000 live birth in Hungary[3]. Autosomal recessive disorder inheritance is based on pedigrees reports with multiple affected siblings but unaffected parents, frequent parental consanguinity, and segregation ratios that suggested primary microcephaly is caused by recessive genes. Molecular basis of primary microcephaly are heterogeneous, suggesting this disorder more frequent in Asian and Arab populations where consanguineous marriage is very common[4]. Molecular changes in the MCPH genes are responsible for the increase in brain size during human evolution[5]. This inherited condition is caused by a genetic defect called mutation and genes associated with this disorder have been found on chromosomes 1, 8, 9, 13, 15 and 19 only[6]. The most recurrent mutations that have been strongly isolated are localized to the gene on chromosome 1 known as ASPM and these mutations impair brain growth and development as it plays a critical role in brain cell division[7].There are total seven MCPH loci and five of the genes have been identified. MCPH1, encoding Microcephalin at 8p23.1 in Northern Pakistan[8]; MCPH3, encoding CDK5RAP2 at 9q33.2 Northern Pakistan[9]; MCPH5, encoding ASPM at 1q31 in Pakistan and Turkey[10]; MCPH6, encoding CENPJ at 13q12.2 in Brazil[11] and MCPH7, encodes STIL at 1p33 in India[12]. MCPH2 at 19q13.1-13.2 in Northern Pakistan[13] while MCPH4 at 15q15-21 in Morocco family[14] showed linkage but their possible gene is still unidentified.Each recessive locus was initially mapped in a large consanguineous family by autozygosity mapping techniques, using panels of ~400 polymorphic autosomal microsatellite markers spread throughout the human genome at a distance of ~12 cM. MCPH5 is the most common locus, accounting for linkage of nearly half of the total affected families and second common locus is MCPH2. MCPH7, MCPH6, MCPH1 and MCPH3 were each found less whereas MCPH4 linkage has not been found in the Pakistani population[11].Exclusion mapping is a technique for narrowing down the location of a gene involved and comparing the chromosomes of affected siblings with an opportunity to map the most likely location of the gene by excluding areas of the chromosome that are not alike[15]. Haplotyping is a combination of alleles at multiple loci that are transmitted together on the same chromosome[16]. Haplotyping data is extremely valuable in the mapping of disease-susceptible genes, particularly in the identification of genes related to inherited disorders but it is limited to small numbers of loci over short distances[17].The aim of the present study was the linkage analysis of three different Pakistani families of autosomal recessive primary microcephaly.
2. Materials and Methods
2.1. Family History
- Before the start of this study mutual consent was obtained from all family members who participated in the study. The data of three families of autosomal recessive primary microcephaly (MCP1, MCP2 and MCP3) was assembled to generate a family history regarding the inheritance and chance occurrence of disease in offspring. The families were studied from population due to socio-ethnic reasons; the consanguineous marriages are common and thus are suitable for locating the defective genes by genetic linkage studies. Three families were collected from Punjab province of Pakistan out of which MCP1 and MCP2 were both Saraiki, while MCP3 was Punjabi. Family MCP1 with seven individuals was collected, out of which two sons were affected. Family MCP2 comprised in three generations so peripheral blood was collected from total twelve individuals out of which four male individuals were affected. MCP3 family was comprised of four individuals with two affected sons while mother was deceased in this family. For genotypic association, clinical manifestations as age, sex, related disorders and most important family history to analyze genetic association were obtained from clinical histories of affected individuals. Complete family history was noted down where subjects were showing mild to moderate mental retardation and sharply slopping forehead and they were not associated with any other abnormality. Affected individuals were having occipito-frontal circumference (OFC) in range of 22-29 cm while forehead to end measurement was from 35-44 cm average varying according to age that was from 4-15 years. The normal individuals from each family were used as control. Peripheral blood samples of microcephalic affected individuals along with their parents and siblings were collected from respective areas and samples were stored in EDTA vials at -20°C.
2.2. Extraction of Genomic DNA
- Genomic DNA of all the affected and normal family members was extracted from peripheral blood lymphocytes by using standard phenol-chloroform method[18].
2.3. PCR Amplification and Genotyping
- PCR amplification of all autosomal markers was performed according to manufacturer specifications. Microsatellite markers used for genotyping included D1S2757, D1S2816, D1S2622, D1S373, D1S1811, D1S1723, D1S2655, D1S1678 (MCPH5), D19S226, D19S416, D19S425, D19S570, D19S881, D19S400, D19S420, D19S418 (MCPH2), D1S2797, D1S2874, D1S197 and D1S417 (MCPH7). D13S787, D13S221, D13S1304 (MCPH6), D9S1872, D9S1682, D9S1881, D9S1821 (MCPH3), D15S98, D15S659, D15S962 (MCPH4), D8S1798, D8S277, D8S1819, D8S1835 (MCPH1).The reaction mixture contained 750 mM Tris-HCl (pH 8.8), 200 mM (NH4) SO4, 2.5 µl of 25 mM MgCl2, 2.0 µ1 of 2.5 mM dNTPs, 1 µl of each 50 pM Primer, 5U/µl Taq DNA polymerase (Fermentas) and 2 µl of DNA extracted from peripheral blood samples. PCR reaction was performed with 5-min initial denaturating step at 94°C; followed by 35 cycles, each cycle comprised denaturation at 94°C for 45-s, annealing at 58°C for 45-s and primer extension at 72°C for 45-s. Final elongation step was carried out at 72°C for 10-min and stored at 4°C until analyzed. PCR amplification of polymorphic microsatellite markers was performed by using 40ng of genomic DNA and amplified markers were pooled and electrophoresed on 8% non-denaturing polyacrylamide gel at 75V for 1 hr. Alleles were numbered according to the CEPH Genotype database V10.0.
2.4. Linkage Analysis
- A fully penetrant autosomal recessive mode of inheritance was assumed and average heterozygosity of the markers in current study was more than 80% that were authentic and highly informative for the allelotyping pedigree members. Two-point analysis was performed by linkage analysis programs like MLINK program of the FASTLINK computer package for fine mapping marker loci.
3. Results
- In our study three Pakistani families (MCP1, MCP2 and MCP3) with autosomal recessive primary microcephaly have been ascertained. The pedigree structure is based upon interviews with multiple family members while personal interviews with key figures in the kindred clarified the consanguineous relationships. The pedigree of each family (MCP1, MCP2 and MCP3) is shown in figure.1A-C respectively, where the consanguineous pedigrees provide convincing evidence of autosomal recessive mode of inheritance. Out of three MCPH families; two got linked at known loci while one family failed to link with any known loci. Microsatellite markers selected from the Marshfield genetic map were according to the prevalence of MCPH so first of all MCP1 was screened with MCPH5 where it showed linkage. MCP1 was screened with a set of total eight markers spanned at a region from 209.15 cM to 218.46 cM. Genotyping of MCP1 shown in figure. 2A and 2B with microsatellite markers D1S373 and D1S1723 respectively.
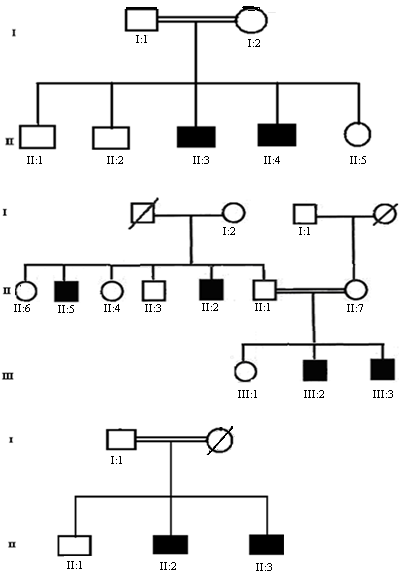 | Figure 1. (A-C). Pedigree of the MCP2 segregating for autosomal recessive primary microcephaly families. A: MCP1 family with seven individuals; B: MCP2 family of twelve individuals and C: MCP3 family having four persons. Circles represent females, squares represent males. Filled circle and squares represent affected individuals |
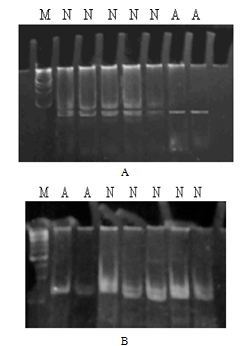 | Figure 2. Electropherogram of ethidium bromide stained 8% non- denaturing polyacrylamide gel of MCP1 on chromosome 1q31 showing homozygosity among all affected (A) while heterozygosity in all normal (N) individuals. A: microsatellite marker D1S373 (214.08cM) and B: microsatellite marker D1S1723 (215.02cM) |
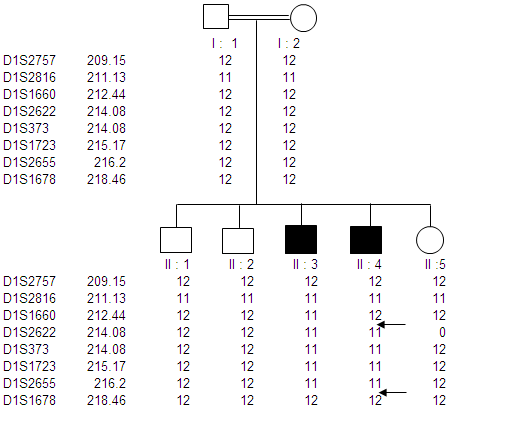 | Figure 3. Pedigree of family MCP1 showing primary microcephaly associated haplotype on chromosome 1q31.1. The genetic map distances according to Marshfield genetic map in centimorgan (cM) are shown next to the markers respectively |
 | Figure 4. Electropherogram of ethidium bromide stained 8% non- denaturing polyacrylamide gel of MCP2 on chromosome 19q13.1 showing homozygosity among all affected (A) and heterozygosity among all normal (N) individuals. A: microsatellite marker D19S425 (59.36cM) and B: microsatellite marker D19S570 (62.03 cM) |
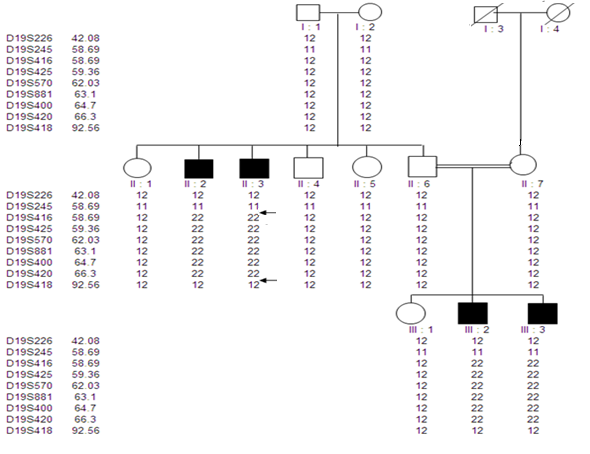 | Figure 5. Pedigree of family MCP2 showing primary microcephaly associated haplotype on chromosome 19q13.1-13.2. The genetic map distances according to Marshfield genetic map in centimorgan (cM) are shown next to the microsatellite markers |
|
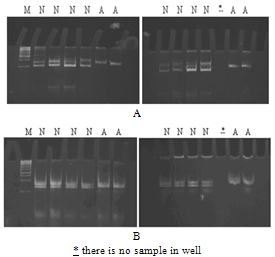
 | Figure 6. Electropherogram of ethidium bromide stained 8% non-denaturing polyacrylamide gel of MCP3 showing heterozygosity among all affected (A) and normal (N) individuals. A: microsatellite marker D1S2622 (214.08cM) on chromosome 1q31 for locus MCPH5; B: microsatellite marker D13S787 (8.87 cM) on chromosome 13q12.2 for locus MCPH6; C: microsatellite marker D19S570 (62.30cM) on chromosome 19q13.1 for locus MCPH2; D: microsatellite marker D1S2874 (75.66cM) on chromosome 1p33 for locus MCPH7; E: microsatellite marker D9S1682 (129.72 cM) on chromosome 9q33.2 for locus MCPH3; F: microsatellite marker D15S962 (43.47cM) on chromosome 15q21 for locus MCPH4; G: microsatellite marker D8S277 (8.34cM) on chromosome 8p23.1 for MCPH1 |
4. Discussion
- In the present study, peripheral blood samples from three families MCP1-MCP3 having autosomal recessive mode of inheritance were collected from geographical isolated areas of the Punjab province of Pakistan. Family history of individuals confirmed that the particular disorder is prevailing as a result of cousin marriage directly might be in first or second generation. MCPH has an incidence of 1:30,000 to 1:250,000 in population while 50% individuals with this genetic disorder have ASPM mutation that is the most common cause of MCPH in Pakistani.Genomic DNA of MCP1 family members were amplified with microsatellite markers of MCPH5 locus; all the affected individuals were homozygous at this locus, while the phenotypicaly normal and carrier individuals were heterozygous. Family MCP1 showed allele frequency of all seven individuals by difference in hetrozygosity and homozygosity of band pattern and thus showed complete linkage on chromosome 1q31. On chromosome 1 there are total 45 genes, out of which ASPM is the candidate gene responsible for autosomal recessive primary microcephaly and found on the long arm (q) of this chromosome[19]. So, we have sequenced exon 3 with five primer sets[10] of ASPM gene but did not found any plausible mutation. Selection of exon 3 was on the basis of its biggest size after exon 18. Nine possible mutations have been identified in exon 3 till to date where seven mutations are due to 1 or 2 base deletions and two mutations are due to transversion of base[20]. There must be a mutation among other 27 exons thus sequencing of remaining exons of ASPM gene is suggestive here.Distribution of ASPM mutations in various functional domains showed that exon 3 and exon 18 are having more chances of all types of mutation which may be a deletion, insertion, missense or nonsense. The most common and frequent sites of mutations are found throughout ASPM gene and uptill now there are total 57 ASPM mutations recorded ([10],[19],[21],[22] and[23]).MCP2 family was screened from MCPH5 primers, where the same heterozygous band pattern was observed in all the samples regardless of affected or being normal so it did not show any linkage. After locus 5 and 6, MCPH2 locus was screened according to prevalence order. The family showed a complete linkage with the markers D19S226 (42.09cM) - D19S (92.56cM) against chromosome region 19q13.1-13.2, where all affected individuals of this family were homozygous and normal individuals were heterozygous in banding pattern. MCP2 family got linked at locus 2 on chromosome 19q13.1-13.2 but the gene for MCPH2 is still not identified while the same result was demonstrated by Robert[13].Human and mouse DYRK1B proteins share 97% sequence identity and sequence analysis has mapped this gene to 19q13.1[24]. Most possible candidate gene for MCPH2 locus is DYRK1A, also called as minibrain related kinase (MIRK). MIRK encods a dual-specificity protein kinase, located in a region specified for MCPH2 on chromosome 19q and its protein product is involved in postembryonic neurogenesis so it is hypothesized that this is might be a plausible gene for MCPH2. Recently it is reported that intragenic 52 kb deletion (37,796,500–37,849,000 bp), affecting last three exons of DYRK1A leads to mental retardation and primary microcephaly[25].Two point linkage analyses of MCP1 and MCP2 were carried out using MLINK of the FASTLINK computer pakckage. For the analysis, an autosomal recessive mode of inhereitance with complete penetrance and a disease allele frequency of 0.001 were assumed[26].
5. Conclusions
- Linkage with any known loci was not shown in MCP3 family, thus the genome wide search for the identification of a novel region for this family would be suggested further. Moreover further investigations are recommended for more molecular elucidations of these three families. Hence, genetic counseling, prenatal diagnosis, gene therapy are proposed as a promising tool for the cure of MCPH phenotype.
ACKNOWLEDGMENTS
- We thank the family members for their invaluable participation and cooperation. This work was supported by the grant from University of the Punjab, Lahore.
References
| [1] | Roberts, E., Hampshire, D. J., Pattison, L., Springell, K., Crow, Y., Jafri, H., Corry, P., Kabani, G., Mannon, J., Rashid, Y., Keen, J., Bond, J., and Woods, C. G., 2002, Autosomal recessive primary microcephaly: an analysis of locus heterogeneity and phenotypic variation., J. Med. Genet., 39, 718-721. |
| [2] | Qazi, Q. H., and Reed, T. E., 1975, A possible major contribution to mental retardation in the general population by the gene for microcephaly., Clin. Genet., 7, 85-90. |
| [3] | Szabo, N., Pap, C., Kobor, J., Svekus, A., Turi, S., and Sztriha, L., 2010, Primary microcephaly in Hungary: epidemiology and clinical features., Acta. Paediatr., 99, 690-693. |
| [4] | Darvish, H., Esmaeeli-Nieh, S., Monajemi, G. B., Mohseni, M., Ghasemi-Firouzabadi, S., Abedini, S. S., Bahman, I., Jamali, P., Azimi, S., Mojahedi, F., Dehghan, A., Shafeghati, Y., Jankhah, A., Falah, M., Soltani Banavandi, M. J., Ghani-Kakhi, M., Garshasbi, M., Rakhshani, F., Naghavi, A., Tzschach, A., Neitzel, H., Ropers, H. H., Kuss, A. W., Behjati, F., Kahrizi, K., and Najmabadi, H., 2010, A clinical and molecular genetic study of 112 Iranian families with primary microcephaly., J. Med. Genet., 47, 823-828. |
| [5] | Woods, C. G., Bond, J., and Enard, W., 2005, Autosomal Recessive Primary Microcephaly (MCPH): A Review of Clinical, Molecular, and Evolutionary Findings., Am. J. Hum. Genet., 76, 717-728. |
| [6] | Kumar, A., Blanton, S. H., Babu, M., Markandaya, M., and Girimaji, S. C., 2004, Genetic analysis of primary microcephaly in Indian families: novel ASPM mutations., Clin. Genet., 66, 341-348. |
| [7] | Pulvers, J. N., Bryk, J., Fish, J. L., Wilsch-Brauninger, M., Arai, Y., Schreier, D., Naumann, R., Helppi, J., Habermann, B., Vogt, J., Nitsch, R., Toth, A., Enard, W., Paabo, S., and Huttner, W. B., 2010, Mutations in mouse Aspm (abnormal spindle-like microcephaly associated) cause not only microcephaly but also major defects in the germline., P.N.A.S., 107, 16595-19600. |
| [8] | Jackson, A. P., McHale, D. P., Campbell, D. A., Jafri, H., Rashid, Y., Mannan, J., Karbani, G., Corry, P., Levene, M. I., Mueller, R. F., Markham, A. F., Lench, N. J., and Woods, C. G., 1998, Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter., Am. J. Hum. Gene., 63, 541-546. |
| [9] | Moynihan, L., Jackson, A. P., Roberts, E., Karbani, G., Lewis, I., Corry, P., Turner, G., Mueller, R. F., Lench, N. J., and Woods, C. G., 2000, A Third Novel Locus for Primary Autosomal Recessive Microcephaly Maps to Chromosome 9q34., Am. J. Hum. Genet., 66, 724-727. |
| [10] | Bond, J., Roberts, E., Mochida, G. H., Hampshire, D. J., Scott, S., Askham, J. M., Springell, K., Mahadevan, M., Crow, Y. J., Markham, A. F., Walsh, C. A., and Woods, C. G., 2002, ASPM is a major determinant of cerebral cortical size., Nat. Genet., 32, 316-320. |
| [11] | Leal, G. F., Roberts, E., Silva, E. O., Costa, S. M. R., Hampshire, D. J., and Woods, C. G., 2003, A novel locus for autosomal recessive primary microcephaly (MCPH6) maps to 13q12.2., J. Med. Genet., 40, 540-542. |
| [12] | Kumar, A., Girimaji, S. C., Duvvari, M. R., and Blanton, S. H., 2009, Mutations in STIL, Encoding a Pericentriolar and Centrosomal Protein, Cause Primary Microcephaly., Am. J. Hum. Gent., 84, 286-290. |
| [13] | Robert, E., Jakson, A. P. Carradice, A. C., Deeble, V. J., Mannan, J., Rashid, Y., Jafri, H., McHale, D. P., Markham, A. F., Lench, N. J., and Woods, C. G., 1999, The second locus for Autosomal recessive primary microcephaly (MCPH2) maps to chromosome 19q13.1-13.2., Eu. J. Hum. Genet., 7, 815-820. |
| [14] | Jamieson, C. R., and Abramowicz, M. J., 1999, Primary Autosomal Recessive Microcephaly: Homozygosity Mapping of MCPH4 to Chromosome 15., Am. J. Hum. Genet., 65, 1465-1469. |
| [15] | Yang, Y. J., Liu, Y. Z , Li, M. X., Lei, S. F., Chen, X. D., Sun, X., and Deng, H. W., 2005, Linkage exclusion analysis of two important chromosomal regions for height., Biochem. Biophys. Res. Commun., 335, 1287-1292. |
| [16] | Gao, G., Allison, D. B., and Hoeschele, I., 2009, Haplotyping Methods for Pedigrees., Hum. Hered., 67, 248-266. |
| [17] | Konfortov, B. A., Bankier, A. T., and Dear, P. H., 2007, An efficient method for multi-locus molecular haplotyping., Nucleic. Acids. Res., 35, 3165. |
| [18] | J. Sambrook. E. F. Fritsch. and T. Maniatis, Molecular Cloning: A Laboratory Manual. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), 1989. |
| [19] | Bond, J., Scott, S., Hampshire, D. J., Springell, K., Corry, P., Abramowicz, M. J., and Mochida, G. H., 2003, Protein-Truncating Mutations in ASPM Cause Variable Reduction in Brain Size., Am. J. Hum. Genet., 73, 1170-1177. |
| [20] | Passemard, S., Titomanlio, L., Elmaleh, M., Afenjar, A., Alessandri, J. L., Andria, G., Billette de Villemeur, T., Boespflug-Tanguy, O., Burglen, L., Del Giudice, E., Guimiot, F., Hyot, C., Isidor, B., Megarbane, A., Moog, U., Odent, S., Hernandez, K., Pouvreau, N., Scala, I., Schaer, M., Gressens, P., Gerard, B., and Verloes, A., 2009, Expanding the clinical and radiological phenotype of primary microcephaly due to ASPM mutations., Neurology., 73, 962-969. |
| [21] | Pichon, B., Vankerckhove, S., Bourrouillou, G., Duprez, L., and Abramowicz, M. J., 2004, A translocation breakpoint disrupts the ASPM gene in a patient with primary microcephaly., Eu. J. Hum. Genet., 12, 419-421. |
| [22] | Gul, A., Hassan, M .J., Mahmood, S., Chen, W., Rahmani, S., Naseer, M. I., Dellefave, L., Muhammad, N., Rafiq, M. A., Ansar, M., Chishti, M. S., Ali, G., Siddique, T., and Ahmad, W., 2006, Genetic studies of autosomal recessive primary microcephaly in 33 pakistani families: novel sequence variants in ASPM gene., Neurogenetics., 7, 12983-12988. |
| [23] | Nicholas, A. K., Swanson, E. A., Cox, J. J., Karbani, G., Malik, S., Springell, K., Hampshire, D., Ahmed, M., Bond, J., DiBenedetto, D., Fichera, M., Romano, C., Dobyns, W. B., and Woods, C. G., 2009, The molecular landscape of ASPM mutations in primary microcephaly., J. Med. Genet., 46, 249-253. |
| [24] | Lee, K., Deng, X., and Friedman, E., 2000, Mirk protein kinase is a mitogen-activated protein kinase substrate that mediates survival of colon cancer cells., Cancer. Res., 60, 3631-3637. |
| [25] | van Bon, B. W., Hoischen, A., Hehir-Kwa, J., de Brouwer, A. P., Ruivenkamp, C., Gijsbers, A. C., Marcelis, C. L., de Leeuw, N., Veltman, J. A., Brunner, H. G., and de Vries, B. B., 2011, Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly., Clin. Genet., 79, 296–299. |
| [26] | Cottingham, R., Indury, R. M., and Schaffer, A. A., 1993, Faster sequential genetic linkage computations., Am. J. Hum. Genet., 53, 252-263. |