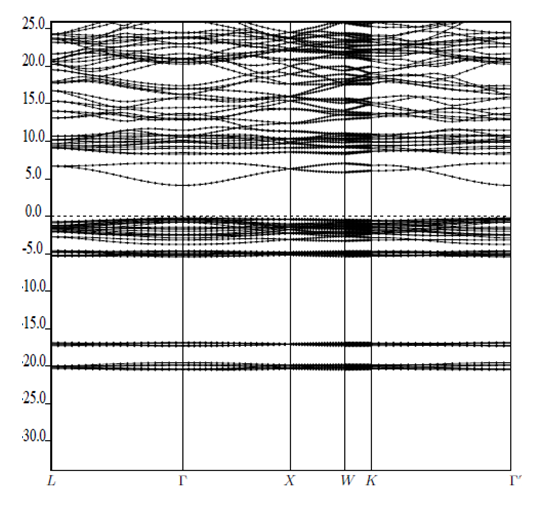
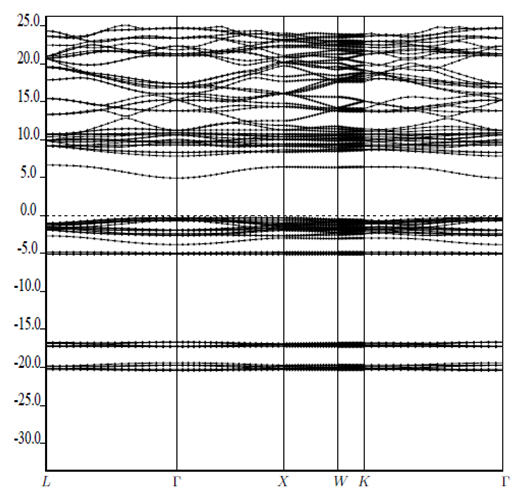
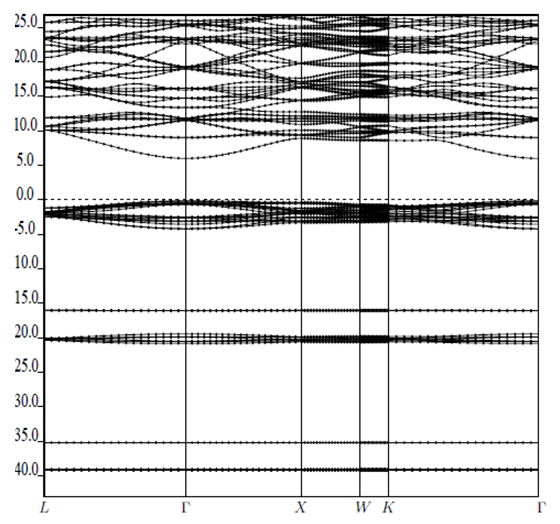
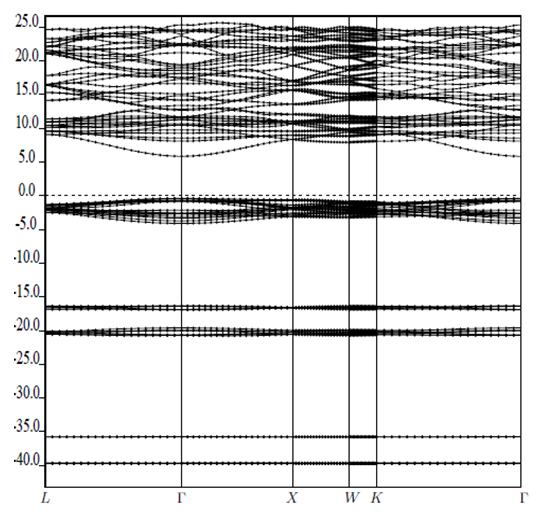
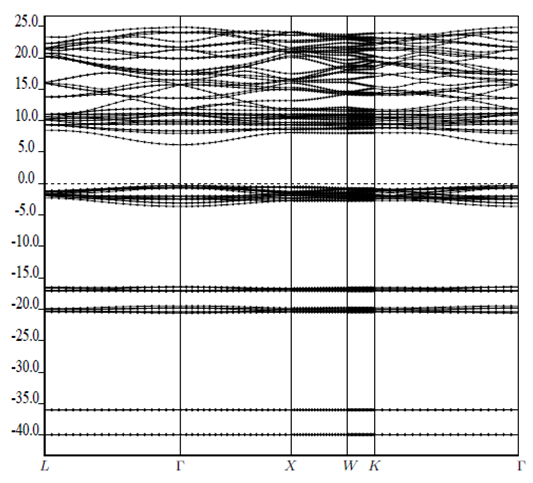
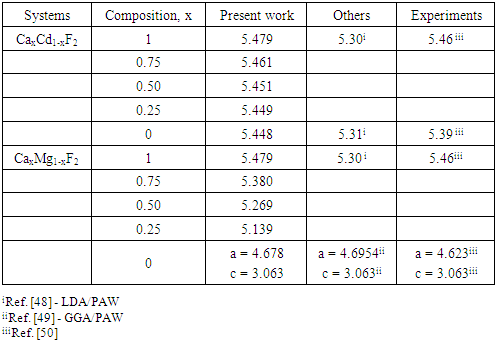
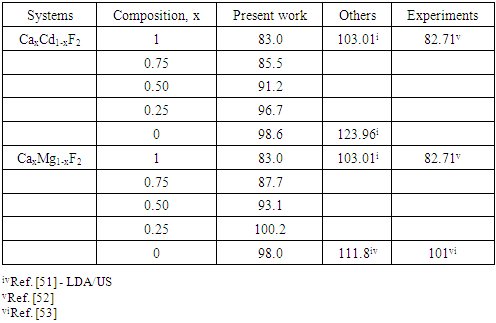
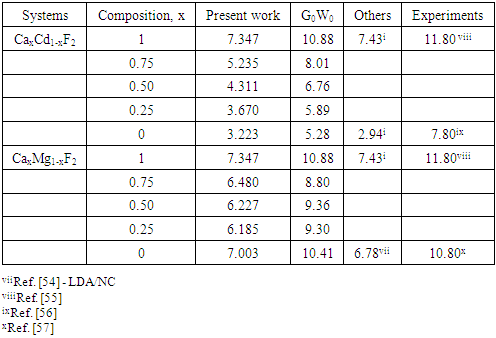
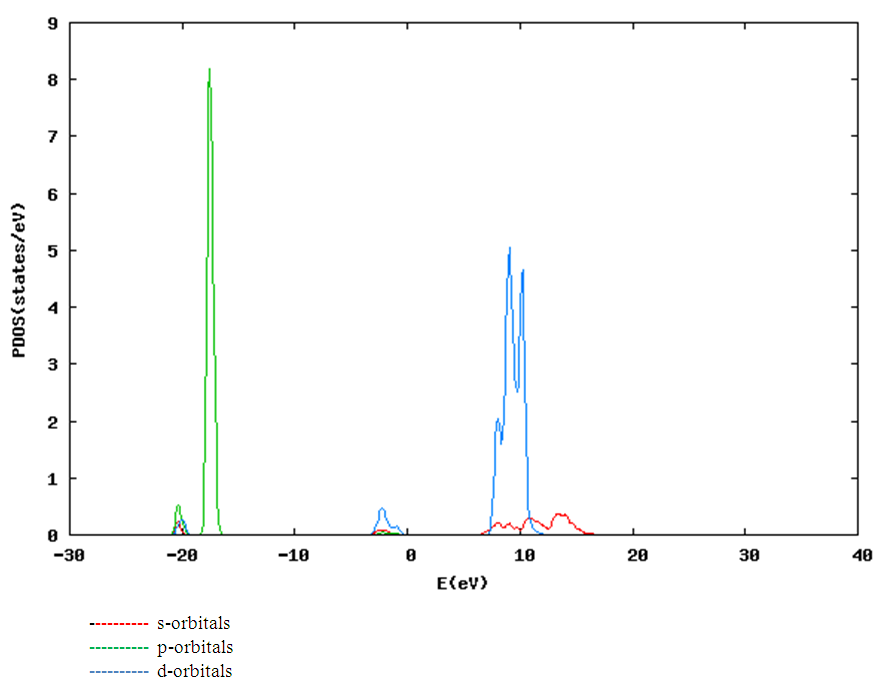
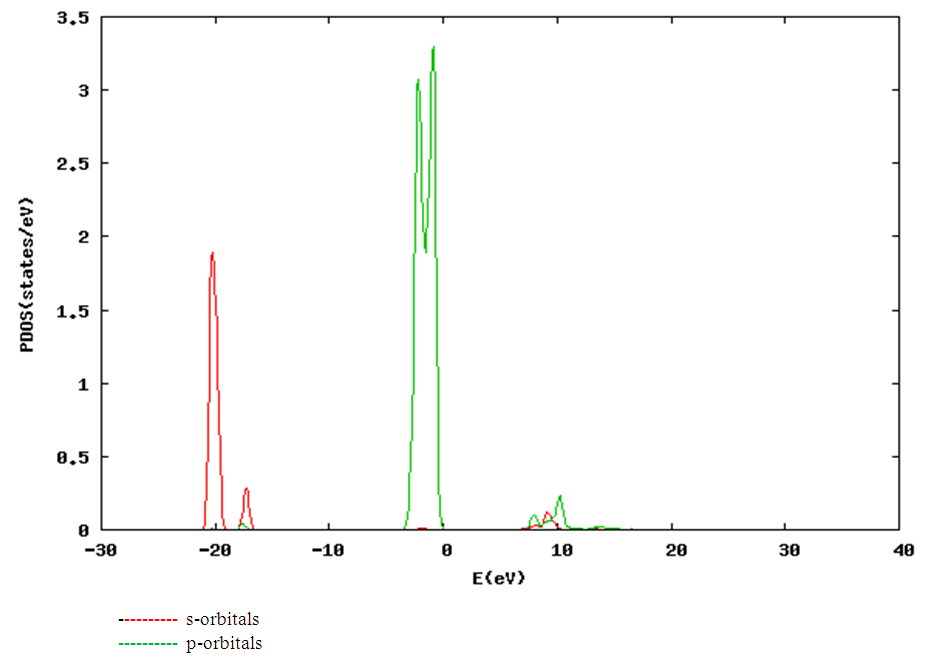
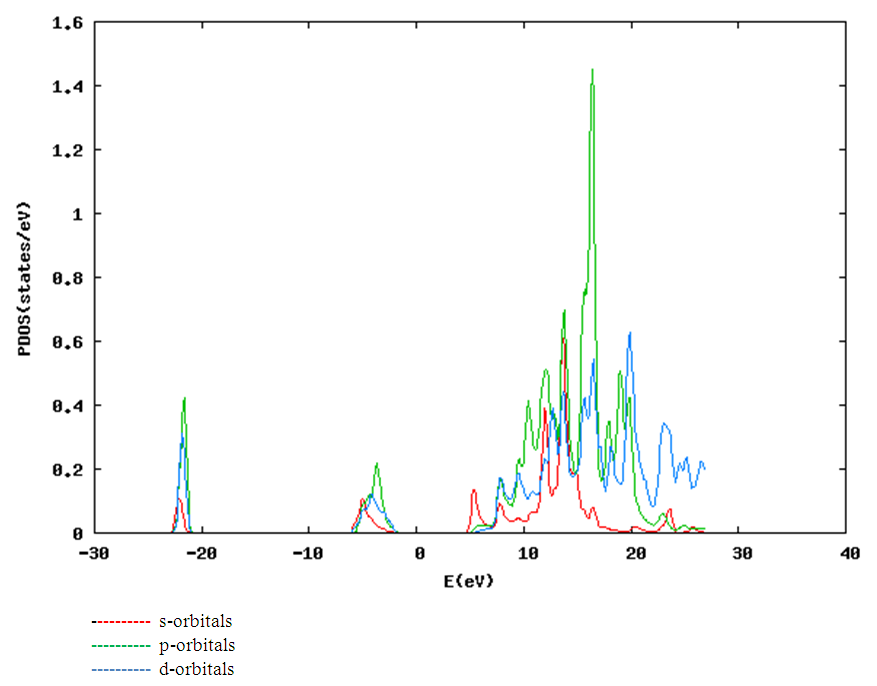
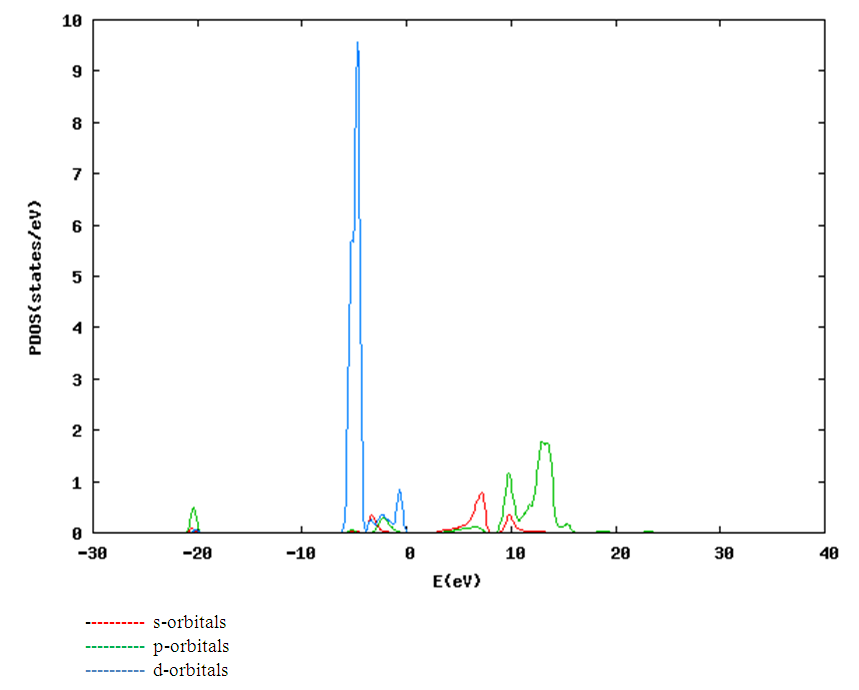
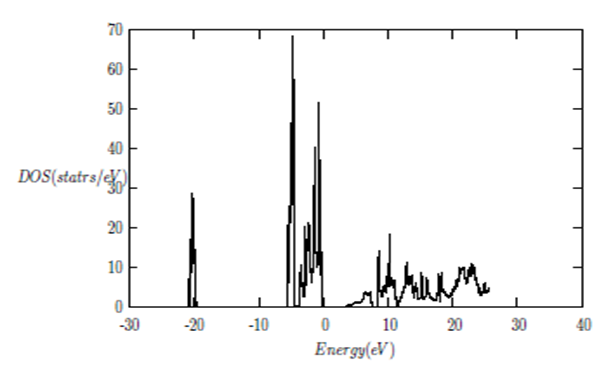
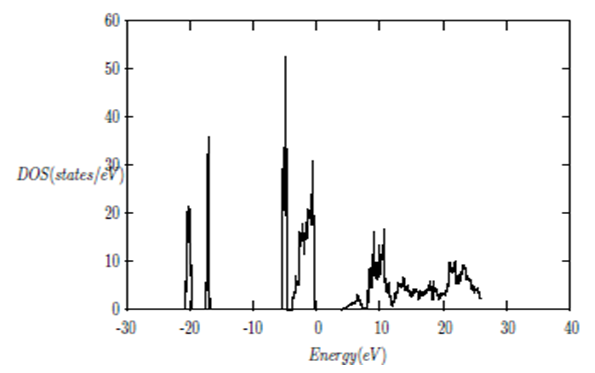
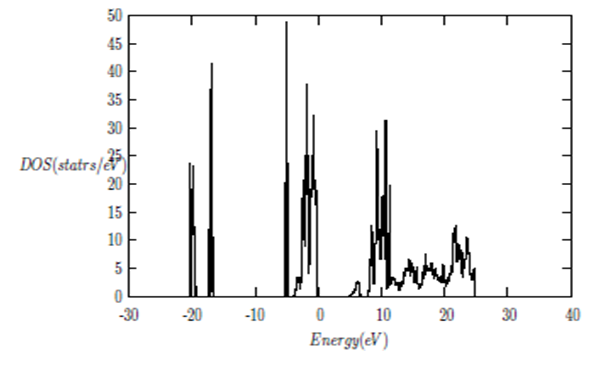
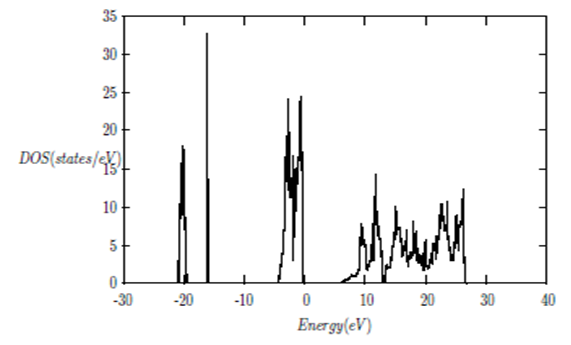
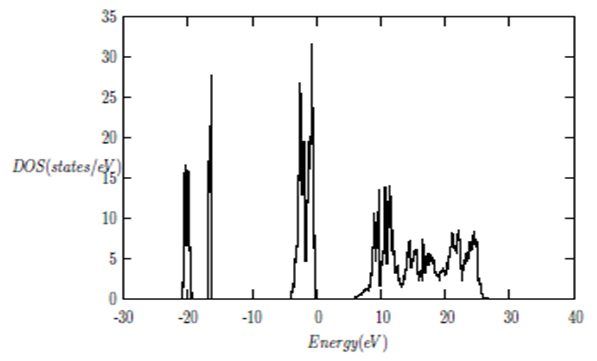
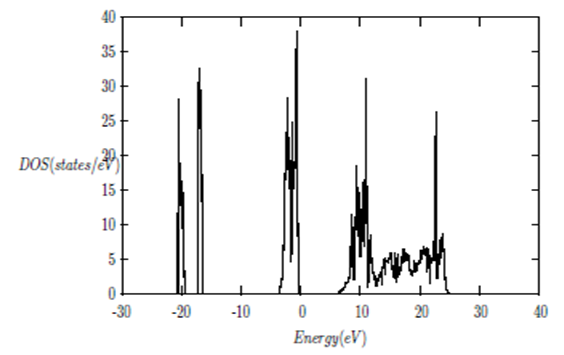
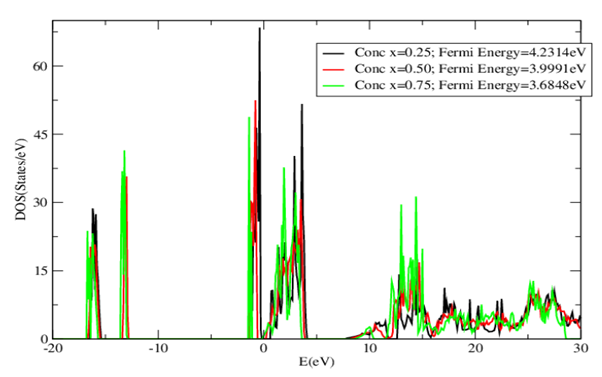
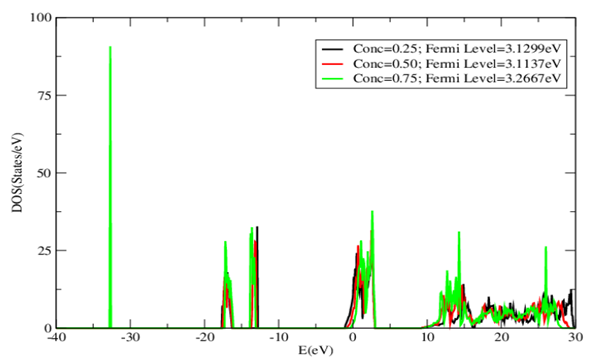
| [1] | K.S. Song, R.T. Williams, “Alkaline Earth Fluorides”, Solid-State Science 105(1993) 96-122. |
| [2] | K. Tsutsui, T. Oshita, S. Watanabe, M. Maeda, “Growth of Fluoride Quantum Well Hetero-structures for Resonant Tunneling Devices on Si Substrates”, ECS Transaction 13(2008) 253-262. |
| [3] | P. Patnaik, Handbook of Inorganic Chemicals, McGraw-Hill, New York, 2003. |
| [4] | O. Duyar, H.Z. Durusoy, “Design and preparation of antireflection and reflection optical coatings”, Turkey Journal of Physics 28(2004) 139-144. |
| [5] | E.L. Shirley, “Many body effects on bandwidths in ionic, noble gas and molecular solids”, Physical Review B 58(1998) 9579-9583. |
| [6] | R. Jia, H. Shi, G. Borstel, “The atomic and electronic structure of CaF2 and BaF2 crystals with H centers: A hybrid DFT calculation study, Journal of Physics: Condensed Matter 22(2010) 055501-055510. |
| [7] | R.A. Evarestov, I.V. Murin, A.V. Petrov, “Electronic structure of fluorite-type crystals”, Journal of Physics: Condensed Matter 1(1988), 6603-6609. |
| [8] | H. Shi, R.I. Eglitis, G. Borstel, “Ab initio calculations of the electronic structure and centers”, Physical Review B 72(2005) 045109-045123. |
| [9] | M. Verstraete, X. Gonze, “First-principles calculation of the electronic, dielectric, and dynamical properties of CaF2”, Physical Review B 68(2003) 195123-195135. |
| [10] | A.S. Foster, C. Barth, A.L. Shluger, R.M. Nieminen, M. Reichling, “Role of tip structure and surface relaxation in atomic resolution dynamic force microscopy: CaF2(111) as a reference surface”, Physical Review B 66(2002) 235-417. |
| [11] | C. Jouanin, C. Gout, “Valence band structure of magnesium fluoride by the tight-binding method”, Journal of Physics C: Solid State Physics 5(1972) 1945-1952. |
| [12] | C. Jouanin, J.P. Albert, C. Gout, “Band structure and optical properties of magnesium fluoride”, Journal of Physics France 37(1976) 595-602. |
| [13] | E. Francisco, J.M. Recio, M.A. Blanco, A.M. Pendas, A. Costales, “The structural and thermodynamical properties of MgF2”, Journal of Physical Chemistry A102(1998) 1595-1601. |
| [14] | V. Kanchana, G. Vaitheswaran, M. Rajagopalan, “High-pressure structural phase transitions in magnesium fluoride studied by electronic structure calculations”, Journal of Alloys and Compounds 352(2003) 60-65. |
| [15] | [A.F. Vassilyeva, R.I. Eglitis, E.A. Kotomin, A.K. Dauletbekova, “Ab initio calculations of MgF2 (001) and (011) surface structure”, Physica B 405(2010) 2125-2127. |
| [16] | A.F. Vassilyeva, R.I. Eglitis, E.A. Kotomin, A.K. Dauletbekova, “Ab initio calculations of the atomic and electronic structure of MgF2 (011) and (111) surfaces”, Central European Journal of Physics 9(2011) 515-518. |
| [17] | A.L. Nikiforov, A.I. Yakimov, S.A. Duarechenskii, S.V. Chaikovskii, “Barrier height and Tunneling current in Schottky diodes with embedded layers of quantum dots”, Journal of experimental and Theoretical Physics Letters 75(2002) 102-106. |
| [18] | H. Shi, R.I. Eglitis, G. Borstel, “Ab initio calculations of the electronic structure and centers”, Physical Review B 72(2005) 045109-045123. |
| [19] | E. Cadelano, G. Cappellini, “Electronic structure of fluorides: general trends for ground and excited state properties”, European Physical Journal 81(2011) 115-120. |
| [20] | G. Cappellini, J. Furthmuller, E. Cadelano, F. Bechstedt, “Electronic and optical properties of Cadmium fluoride: The role of many-body effects”, Physical Review B 87(2011) 075203-075211. |
| [21] | A.I. Kalugin, V.V. Sobolev, “Electronic structure of cadmium fluoride”, Physical Review B 71(2005) 115112-115118. |
| [22] | E. Deligoz, K. Colakoglu, Y. Ciftci, “Elastic, Electronic and Lattice dynamical properties of CdS, CdSe and CdTe”, Physica B: Physics of Condensed Matter 373(2006) 124-130. |
| [23] | J. Haines, J.M. Leger, F. Gorelli, D.D. Klug, J.S. Tse, Z.Q. Li, “X-ray diffraction and theoretical studies of the high-pressure structures and phase transitions in magnesium fluoride”, Physical Review B 64(2001) 134110-134119. |
| [24] | S. Siskos, C. Fountaine, A. Munoz-Yague, “Epitaxial growth of lattice-matched CaxSr1-xF2 on (100) and (110) GaAs substrates”, Journal of Applied Physics 56(1984) 1642-1646. |
| [25] | T. Gotoh, H. Kambayashi, K. Tsutsui, “Epitaxial Growth of CaxCd1-xF2 Mixed Crystal Films on Si Substrates”, Japanese Journal of Applied Physics 39(2000) 476-478. |
| [26] | S. Maeda, N. Matsudo, S. Watanabe, K. Tsutsui, “Crystalline structure of epitaxial CaxMg1-xF2 alloys on Si(100) and (111) substrates”, Thin Solid Films 515(2006) 448-451. |
| [27] | K. Tsutsui, T. Oshita, S. Watanabe, M. Maeda, “Growth of Fluoride Quantum Well Heterostructures for Resonant Tunneling Devices on Si Substrates”, ECS Transactions 13(2008) 253-262. |
| [28] | K. Tsutsui, N. Matsudo, M. Maeda, S. Watanabe, “Resonant Tunneling Diodes on Si Substrates Using Fluoride Heterostructures and Feasibility of Application to Integrated Circuits”, Solid-State Device Research Conference, Proceeding of the 36th European, 439-442, 2006. |
| [29] | A. Izumi, N. Matsbara, Y. Kushida, K. Tsutsui, N.S. Sokolov, “CdF2/CaF2 Resonant Tunneling Diode Fabricated on Si(111)”, Japan. Journal of Applied Physics 36(1997) 1849-1852. |
| [30] | P. Hohenberg, W. Kohn, “Inhomogenous electron gas”, Physical Review B 136(1964) 864-871. |
| [31] | W. Kohn, L.J. Sham, “Self-consistent equations including exchange and correlation effects”, Physical Review A 140(1965) 1133-1138. |
| [32] | J. Perdew, Y. Wang, “Accurate and simple analytic representation of the electron-gas correlation energy”, Physical Review B 45(1992) 13244-13252. |
| [33] | X. Gonze, R. Stumpf, M. Scheffler, “Analysis of separable potentials”, Physical Review B 44(1991) 8503-8513. |
| [34] | H.J. Monkhorst, J.D. Pack, “Special points for Brillouin-zone integrations”, Physical Review B 13(1976) 5188-5192. |
| [35] | F.D. Murnaghan, “The Compressibility of Media under Extreme Pressures”, Proceedings of the National Academy of Sciences of the United States of America 30(1944) 244-247. |
| [36] | C. Rostgaard, K.W. Jacobsen, K.S. Thygesen, “Fully self-consistent GW calculations for molecules”, Physical Review B 81(2010) 085103-085113. |
| [37] | F. Aryasetiawan, O. Gunnarsson, “The GW Method” Reports on Progress in Physics 61(1998) 237-312. |
| [38] | G. Onida, L. Reining, A. Rubio, “Electronic excitations: density-functional versus many-body Green’s-function approaches” Reviews of Modern Physics 74(2002) 601-659. |
| [39] | W. G. Aulbur, L. Jonsson, J. W. Wilkins, “Quasiparticle Calculations in Solids”, Solid State Physics Advanced Research Applications 54(2000) 1-218. |
| [40] | S. Maeda, N. Matsudo, S. Watanabe, K. Tsutsui, “Growth Characteristics of ultrathin epitaxial CaxMg1-xF2 alloys on (111) substrates”, Journal of Crystal Growth 285(2005) 572-578. |
| [41] | G.A. Asha, A. Kumar, M.S. Hegde, U.V. Waghmare, “Structure of Ce1-xSnxO2 and its relation to oxygen storage property from first-principles analysis”, Journal of Chemical Physics 132(2010) 194702-194709. |
| [42] | L. Vegard, “The constitution of mixed crystals and the space occupied by atoms”, Zeitschrift fur Physik B: Condensed Matter 5(1921) 17-26. |
| [43] | F. El Haj Hassan, H. Akbarzadeh, “First-principles elastic and bonding properties of barium chalcogenides”, Computational Material. Science 35(2006) 423-445. |
| [44] | F. El Haj Hassan, S.J. Hashemifar, H. Akbarzadeh, “Density functional study of Zn1−xMgxSeyTe1−y Quaternary semiconductor alloys”, Physical Review B 73(2006) 195-202. |
| [45] | F.E.H. Hassan, S.J. Hashemifar, H. Akbarzadeh, “Density functional study of Zn1−xMgxSeyTe1−y Quaternary semiconductor alloys”, Physical Review B 73(2006) 195-202. |
| [46] | N. Boukhris, H. Meradji, S. Ghemid, S. Drablia, F.E.H. Hassan, “Ab initio study of the structural, electronic and thermodynamic properties of PbSe1-xSx, PbSe1-xTex and PbS1-xTex ternary Alloys”, Physica Scripta 83(2011) 065701-065709. |
| [47] | S. Labidi, M. Labidi, H. Meradji, S. Ghemid, F.E.H. Hassan, “First principles calculations of structural, electronic, optical and thermodyamic properties of PbS, SrS and their ternary alloys Pb1-xSrxS”, Computational Materials Science 50(2011) 1077-1082. |
| [48] | E. Cadelano, G. Cappellini, “Electronic structure of fluorides: general trends for ground and excited state properties”, European Physical Journal 81(2011) 115-120. |
| [49] | R. Golesorkhtabar, P. Pavone, J. Spitaler, P. Puschnig, C. Draxl, “ELaStic: A tool for calculating second-ordered elastic constants from first principles”, Computer Physics Communications 184(2013) 1861-1873. |
| [50] | A.R. West, Basic Solid State Chemistry, John Wiley and Sons, Chichester, England, 1999. |
| [51] | K. Umemoto, R.M. Wentzcovich, D.J. Weidner, J.B. Parise, “NaMgF3: A low-pressure analog of MgSiO3”, Geophysical Research Letter 33(2006) 15304-15307. |
| [52] | R.C. Weast, Chemical Rubber Company Handbook of Chemistry and Physics, CRC Press, Boca Raton, Florida, United States, 1976. |
| [53] | J. Haines, J.M. Leger, F. Gorelli, D.D. Klug, J.S. Tse, Z.Q. Li, “X-ray diffraction and theoretical studies of the high-pressure structures and phase transitions in magnesium fluoride”, Physical Review B 64(2001) 134110-134119. |
| [54] | Y. Zhijun, R. Jia, “Quasi-particle band structures and optical properties of magnesium fluoride”, Journal of Physics: Condensed Matter 24(2012) 085602-085606. |
| [55] | G.W. Rubloff, “Far-Ultraviolet Reflectance Spectra and the Electronic Structure of Ionic Crystals”, Physical Review B 5(1972) 662-684. |
| [56] | B.A. Orlowski, P. Plenkiewicz, “Electronic Band Structure of CdF2: Hotoemission Experiment and Pseudopotential Calculations”, Physical Status Solid B 126(1984) 285-292. |
| [57] | A.T. Davidson, A.M.J. Raphuthi, J.D. Comins, T.E. Derry, “On the nature and efficiency of colouration of MgF2 crystals by 100keV ions”, Nuclear Instrumental Methods B 80/81(1993) 1237-1240. |

 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML