-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Materials Science
p-ISSN: 2162-9382 e-ISSN: 2162-8424
2017; 7(2): 41-45
doi:10.5923/j.materials.20170702.03

Study of Group Charges, Molecular Conformations, and Electrochemical Properties of Nematogens - A DFT Approach
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLSeema Prasad, Durga P. Ojha
School of Physics, Sambalpur University, Jyoti Vihar, Sambalpur, Odisha, India
Correspondence to: Durga P. Ojha, School of Physics, Sambalpur University, Jyoti Vihar, Sambalpur, Odisha, India.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

In the present article, structure of nematogenic p-n-Alkoxycinnamic acids (nOCAC) with alkyl chain carbon atoms (n=6, 8) has been optimized using the density functional Becke3-Lee-Yang-Parr (B3LYP) exchange-correlation with 6-31+G (d) basis set using crystallographic geometry as input. The dimer complexes of higher homologues (n=6, 8) during the different modes of interactions, and their properties have been reported based on Density Functional Theory (DFT) calculations. The electrochemical properties have been investigated. The phase stability of these nematogens has been analyzed based on Mulliken and Loewdin population analysis.
Keywords: Nematogen, Phase stability, Electrochemical properties
Cite this paper: Seema Prasad, Durga P. Ojha, Study of Group Charges, Molecular Conformations, and Electrochemical Properties of Nematogens - A DFT Approach, American Journal of Materials Science, Vol. 7 No. 2, 2017, pp. 41-45. doi: 10.5923/j.materials.20170702.03.
Article Outline
1. Introduction
- Liquid Crystals (LCs) materials are of fundamental interest for a variety of applications in the areas of optical signal processing, and photo-induced phenomena [1]. In recent years, a field of research that is growing steadily is the photo-induced phenomenon in which the wavelengths of incident light impinge on the material stability [2]. Photo stability of liquid crystal (LC) materials plays a crucial role in affecting the lifetime of liquid crystal display (LCD), and memory devices.Liquid Crystals (LCs), as the crystals that flow, have gained much prominence in multidirectional facts with realistic models [3, 4] and excited practical applications. The foremost focus is to explore molecular chemistry [5, 6] in order to study their practicability in technological applications. The design and synthesis of new organic compounds, which exhibit a nematic phase with a specific spectrum of properties, have essential contributions in establishing and expanding the multibillion dollar LCD industry. The need to develop new liquid crystalline (LC) compound, for desired applications, and technology provides a new motivation to develop a deeper understanding in molecular electronic structure and bonding [7, 8]. LC behavior is a genuine supramolecular phenomenon based on the existence of extended weak interactions (dipole-dipole, dispersion forces) between the molecules. It is usually necessary that these molecules have anisotropic shapes [9] able to pack efficiently, so that these weak interactions can be high in number, co-operate, and become strong as to keep the molecules associated in a preferred orientation. The differences between the nematic behavior of simple conventional mesogens (monomers) and dimers are thought to stem entirely from the configurational correlations [10] that the spacers impose on the mesogenic units. Thus, the inclusion of such correlations constitutes the major step in the formulation of a molecular theory that goes from the description of aggregation behavior of single mesogen to dimer, and to multi-mesogen phases. The methods based on Time Dependent Density Functional Theory (TDDFT) applied to small and middle sized systems [11, 12] provide rather good accuracy at low computational cost [13, 14]. These methods still remain of rather limited application for establishing realistic molecular models and biological systems. The present theoretical approach aims at providing a comparative picture of higher homologues nematogenic p-n-Alkoxycinnamic acids (nOCAC, n= 6, 8) using the DFT method [13]. The electrochemical properties such as HOMO (EH), LUMO (EL) energies, energy gap (Eg), ionization energy (I), electron affinity (A), electro negativity (χ), chemical hardness (η), electronic chemical potential (μ), electrophilicity index (ω), and softness (S) have been investigated. The dimer complexes during the different modes of interactions and their properties have been reported based on DFT calculations. An attempt has been made to find out the most energetically favourable configuration in each mode of interaction. An examination of thermodynamic data has revealed that nOCAC molecules exhibits nematic-isotropic transition temperature as follows [15]: 6OCAC at 452K; and 8OCAC at 445K.
2. Computational Method
- The main difficulties against a reliable theoretical approach are concerned with the size of such systems, and the presence of strong electron correlation effects. Both properties are difficult to treat in the framework of the quantum mechanical methods rooted in the Hatree- Fock (HF) theory. Density functional theory (DFT) is successful to evaluate a variety of ground- state properties with accuracy close to that of post-HF methods [13, 16]. In this context, remarkable structural predictions have been obtained especially using the ‘hybrid’ density functional [17-19] such as B3LYP combining ‘exact exchange’ with gradient-corrected density functionals. The present model comprises of the monomer and dimer assemblies of p-n-Alkoxycinnamic acids (nOCAC, n=6, 8). The geometry optimizations have been performed using the density functional theory (DFT) approach [13] and in particular the Becke3-Lee-Yang-Parr hybrid functional (B3LYP) exchange-correlation with 6-31G+ (d) basis set. The DFT approach was originally developed by Hohenberg and Kohn [20], Kohn and Sham [21, 22] to provide an efficient method of handling many-electron system. The theory allows us to reduce the problem of an interacting many-electron system to an effective single-electron problem. The DFT calculations have been performed by a spectroscopy oriented configuration interaction procedure (SORCI) [23], whereas, a revised version of QCPE 174 by Jeff Reimers, University of Sydney and coworkers have been used for the semiempirical calculations [24].The general structural parameters of the systems such as the standard values of bond lengths and bond angles have been taken from published crystallographic data [15]. The charge distributions of the molecules have been calculated by performing Mulliken and Loewdin population analysis. The mesomorphic behavior and the nematic phase stability have been predicted through the calculated local charge distributions.
3. Results and Discussion
- The electronic structures of nOCAC (n=6, 8) have been shown in Fig. 1. The structures have been constructed on the basis of published crystallographic data with standard values of bond lengths and bond angles [15]. Generally, the trans isomers are linear in shape and tend to form mesophases, while the cis isomers are not linear, and tend not to form mesophases. The structures of the systems have the all-trans extended conformation, and the molecules exist in the crystals as planar hydrogen-bonded dimers. The molecular charge distribution, and phase stability of the molecules has been analyzed as given below:
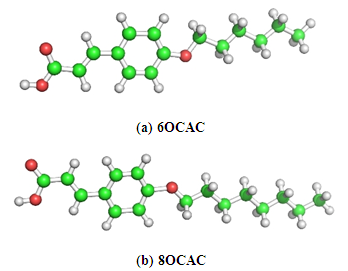 | Figure 1. The electronic structures of (a) 6OCAC, and (b) 8OCAC molecules |
3.1. Group Charges
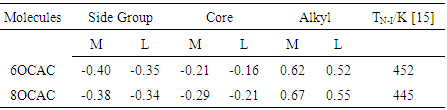
- The group charges are needed to explain the behavior of mesogens. Mulliken population analysis, which partitions the total charge among the atoms in the molecule, has been performed, and the results have been compared with those obtained from Loewdin population analysis. However, there is much agreement among the methods while it comes to the group charges of each molecule. It is evident from (Table 1) that the positively charged alkyl chains of 6OCAC will be strongly attracted by the negatively charged side group as well as the core, causing the formation of longer units in the nematic phase. Hence, the nematic phase stability is expected to be high for 6OCAC. Further, the thermal vibration amplitude of the chain carbon atoms increase markedly with the increase of chain length, indicating a low packing efficiency for higher homologues. This leads to the drastic decrease in nematic phase stability. This is in agreement with the nematic-isotropic transition temperature reported by the crystallographer (Table 1).
|
3.2. Electrochemical Properties
- The HOMO (EH), LUMO (EL) energies, and energy gap (Eg) have an effect on the stability of a molecule. The energy of the HOMO is directly related to the ionization energy, LUMO energy is directly related to the electron affinity. The energy gap between the HOMO, and LUMO is an important factor in the determination of molecular transport properties and photo stability. The chemical hardness is a measure for resistance to deformation or change, is very important tool to study the stability of molecular systems, and is also an approximation to the first electron excitation energy. The lowering of energy separation between the HOMO and LUMO clearly explicates the charge transfer interactions taking place within the molecule. The average value of the HOMO and LUMO energies is related to the electro negativity. The negative of the electro negativity is the chemical potential (μ). A comparative picture of electrochemical properties such as HOMO (EH), LUMO (EL) energies, the energy gap (Eg), ionization energy (I), electron affinity (A), chemical hardness (η), electronic chemical potential (μ), electrophilicity index (ω), and softness (S) of isolated molecules have been reported in Table 2 (a, b). Evidently, the individual stacked dimers of 8OCAC exhibit lower band; hence its conductivity is high in comparison with 6OCAC. Further, the same trend has been observed from the in-plane, and terminal dimers.
|
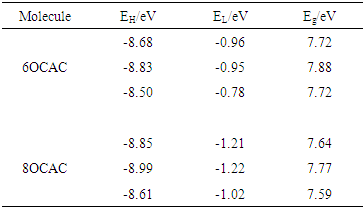
3.3. Conformational Behavior of Molecules
- The results obtained through these computations suggest that the optical properties of isolated molecules are influenced by their electronic structures. Therefore, in order to understand the self organizing ability of mesogens, the different modes of molecular interactions namely; stacking, in-plane, and terminal interactions have been considered between a pair of nOCAC (n=6, 8) molecules. The interaction energies of dimer complexes have been taken to investigate the most energetically stable configuration using the DFT method. The basic idea underlying the study of molecular conformations is to study the physical and chemical properties of compounds that are closely related to the preferred conformations. The conformational behavior of LCs displays a large variation in intermolecular effects that depends on the nature and magnitude of interactions. Each conformation may exhibit the distinct energy, and lower energy conformations will be populated in preference to those of higher energy. The most energetically stable configurations of 6OCAC and 8OCAC molecules have been shown in Fig. 2 and Fig. 3 respectively. A comparison of stacked dimers between both the molecules suggests that the extension of the chain length to eight carbon atoms, a recognizable segregation of the dimers into a highly tilted layer structure has been obtained. The mutual interaction between the dimers in the structure is, however, quite weak, in particular to chain atoms. Hence, the end chains provide enough disorder to the crystal to pass on to nematic rather than smectic phase. The end chains have the all-trans extended conformation, and molecules exist in the crystal as planar hydrogen-bonded dimers. The dimers are arranged in end-to-end fashion in parallel rows. Similarly, side-to-side packing of dimers (Fig. 2b & Fig. 3b) is found in each molecule in a good fit between adjacent aromatic cores. But the amplitudes of thermal vibration of the carbon chain atoms increase markedly with the increase of chain length, indicating a low packing efficiency, and low nematic-isotropic transition temperatures that is in agreement with charge distribution analysis reported in this article.
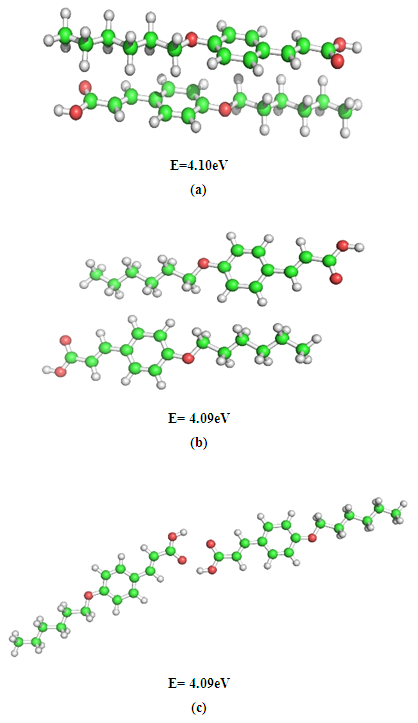 | Figure 2. Energetically favourable structures of 6OCAC dimer in (a) stacking, (b) in-plane, and (c) terminal interactions |
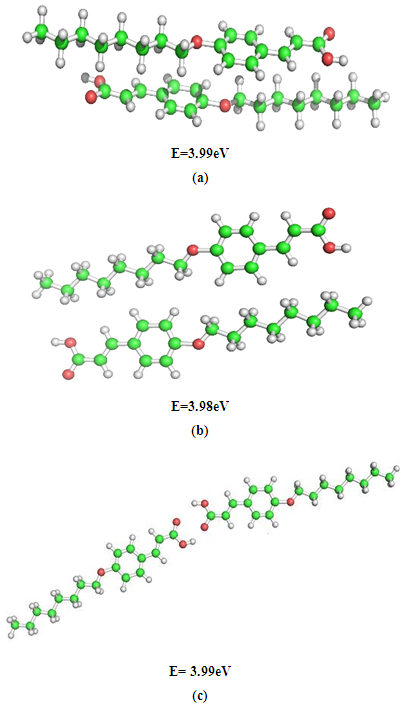 | Figure 3. Energetically favourable structures of 8OCAC dimer in (a) stacking, (b) in-plane, and (c) terminal interactions |
4. Conclusions
- 1. A comparison of stacked dimers of nOCAC (n=6, 8) molecules suggests that the extension of the chain length to eight carbon atoms, a recognizable segregation of the dimers into a highly tilted layer structure has been observed. The mutual interaction between the dimers in the structure is quite weak, in particular to chain atoms. Hence, the end chains provide enough disorder to the crystal to pass on to nematic rather than smectic phase. 2. The energy gap (Eg) values help us to determine the molecular reactivity such as the ability to absorb light, and to react with other species, a molecule with small gap is expected to have higher reactivity and lower stability in photo-physical process. However, the energy gap shows a preference with increment in the end alkyl groups. The increment of alkyl groups in the end chain cause a decrement in energy gap, thereby increasing the conductivity of the molecule. 3. The increase of chain length, indicating a low packing efficiency for higher homologues. This leads to the drastic decrease in nematic phase stability.
ACKNOWLEDGEMENTS
- One of the authors, Seema Prasad, is thankful to the UGC, New Delhi, India for providing financial support as JRF under the RGNF scheme.
References
| [1] | M. Vijaysrinivasan, P. Kannan, A. Roy, “Dual switchable six-ring bent- core liquid crystals with azo linkages exhibiting B1 and B2 mesophases,” Liq. Cryst., 39, 1465- 1475 (2012). |
| [2] | A. Ramamoorthy, Thermotropic liquid crystals: Recent advances, Springer, Germany (2007). |
| [3] | P. L. Praveen, and D. P. Ojha, “Calculation of spectral shift in UV-Visible region and photoresponsive behavior of Fluorinated liquid crystals,” Mat. Chem. Phys., 135, 628- 634 (2012). |
| [4] | P. L. Praveen, and D. P. Ojha, “Influence of alkyl chain length and solvents on configurational probability of liquid crystalline materials,” Mol. Cryst. Liq. Cryst., 608, 72- 81 (2015). |
| [5] | J. J. Travadi, M. S. Vadodaria, K. D. Ladva, and A. V. Doshi, “Mesomorphism dependence on molecular structure,” Mol. Cryst. Liq. Cryst., 626, 21- 30 (2016). |
| [6] | T. N. Govindaiah, “Optical studies on Smectic-Isotropic phase transition behavior of two thermotropic liquid crystals,” Mol. Cryst. Liq. Cryst., 626, 108- 114 (2016). |
| [7] | Z. Zhang, and H. Guo, “The phase behavior, structure, and dynamics of rodlike mesogens with various flexibility using dissipative particle dynamics simulation,” J. Chem. Phys., 133, 144911-144923 (2010). |
| [8] | M. V. Gorkunov, M. A. Osipov, A. Kocot, J. K. Vij, “Molecular model of biaxial ordering in nematic liquid crystals composed of flat molecules with four mesogenic groups,” Phys. Rev. E 81, 061702- 061712 (2010). |
| [9] | S. D. Peroukidis, A. G. Vanakaras, D. J. Photinos, “Molecular modeling of liquid crystalline self-organization of Fullerodendrimers: Columnar to Lamellar phase transitions driven by temperature and/or concentration changes,” J. Phy. Chem. B, 112, 12761- 12767 (2008). |
| [10] | D. P. Ojha, “A Computational analysis of ordering in ABCHN at Nematic-Isotropic transition temperature,” J. Mol. Model, 12, 161- 167 (2006). |
| [11] | G. Bringmann, K. Maksimenka, J. M. Comar, M. Knauer, T. Bruhn, “The absolute axial configurations of knipholone and knipholone anthrone by TDDFT and DFT/MRCI CD calculations: A Revision,” Tetrahedron, 63, 9810- 9824 (2007). |
| [12] | M. Pecul, K. Ruud, T. Helgaker, “Density functional theory calculation of electronic circular dichroism using London orbitals,” Chem. Phys. Lett., 388, 110- 119 (2004). |
| [13] | W. Koch, M. C. Holthausen, A Chemist’s Guide to Density Functional Theory; Wiley- VCH: Weinheim (2000). |
| [14] | H. L. Peng, J. L. Payton, J. D. Protasiewicz, M. C. Simpson, “Twisting the Phenyls in Aryl Diphosphenes (Ar-P=P-Ar) significant impact upon lowest energy excited states,” J. Phys. Chem. A, 113, 7054- 7063 (2009). |
| [15] | R. F. Bryan, P. Hartley, “An X– Ray study of the p-n-Alkoxycinnamic acids. Part III. Crystal structures of four nematogenic acids having two, four, six, and eight alkyl chain carbon atoms,” Mol. Cryst. Liq. Cryst., 69, 47- 69 (1981). |
| [16] | A. Neugebauer, G. Hafelinge, “Reliability of ab-initio methods and basis set dependencies for accurate prediction of re distances of CO bond lengths,” J. Mol. Struct. Theochem., 585, 35- 47 (2002). |
| [17] | K. Sharkas, J. Toulouse, A. Savin, “Double-hybrid density-functional theory,” J. Chem. Phys. 134, 064113- 064122 (2011). |
| [18] | K. Sharkas, A. Savin, H. J. A. Jensen, J. Toulouse, “A multiconfigurational hybrid density-functional theory,” J. Chem. Phys. 137, 044104- 044114 (2012). |
| [19] | Y. Cornaton, O. Franck, A. M. Teale, E. Fromager, “Analysis of double hybrid density functional along the adiabatic connection,” Mol. Phys. 111, 1275- 1294 (2013). |
| [20] | P. Hohenberg, W. Kohn, “Inhomogeneous electron gas,” Phys. Rev., 136, B864 –B871 (1965). |
| [21] | W. Kohn, L. J. Sham, “Self-consistent equations including exchange and correlation effects,” Phys. Rev., 140, A1133- A1188 (1965). |
| [22] | R. O. Jones, O. Gunnarsson, “The density functional formalism, its application and prospects,” Rev. Mod. Phys., 61, 689- 746 (1989). |
| [23] | F. A. Neese, “A spectroscopy oriented configuration interaction procedure,” J. Chem. Phys., 119, 9428- 9444 (2003). |
| [24] | Tejerina, B, Reimers, J, DOI: 10254/nanohub-r3352.9 |