-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Materials Science
p-ISSN: 2162-9382 e-ISSN: 2162-8424
2017; 7(2): 35-40
doi:10.5923/j.materials.20170702.02

Theoretical and Experimental Studies on Liquid Crystals- The Role of Position of Oxygen
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLSeema Prasad, Durga P. Ojha
School of Physics, Sambalpur University, Jyoti Vihar, Sambalpur, Odisha, India
Correspondence to: Durga P. Ojha, School of Physics, Sambalpur University, Jyoti Vihar, Sambalpur, Odisha, India.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

The present article deals with the theoretical and experimental studies on novel liquid crystals. Theoretical studies on nematic liquid crystals viz. 4-octylphenyl-6-octyloxy-2-naphthoate (NAPHE1), and 6-octyloxy-2-naphthyl-4-octyloxybenzoate (NAPHE2) using the density functional theory (DFT) method has been carried out. The structural and electrochemical properties such as HOMO, LUMO energies, and energy gap (Eg) have been investigated. It has been observed that the energy gap is increased due to the substitution of oxygen. Further, the four Schiff’s base compounds are synthesized and characterized by using TM and DSC.
Keywords: Liquid crystals, DFT method, TM, DSC
Cite this paper: Seema Prasad, Durga P. Ojha, Theoretical and Experimental Studies on Liquid Crystals- The Role of Position of Oxygen, American Journal of Materials Science, Vol. 7 No. 2, 2017, pp. 35-40. doi: 10.5923/j.materials.20170702.02.
Article Outline
1. Introduction
- Liquid Crystals (LCs) are soft condensed matter and now commonplace in displays devices, light modulator, temperature sensor and optical communication networks [1]. These demands are being met by the continuous advances [2] in the theoretical knowledge and the software technologies. This provides an informative database for further computations, and support to the experimental observations [3]. They have the different phases like nematic, smectic and cholesteric are innovative class of functional materials and have attracted the great deal of attention due to their various applications in electro-optics. The liquid crystal characteristics need to be satisfied for many applications such as stability of mesophase range, and existence of phases at a desired temperature [4]. The terminal groups present in a molecule are finding an increasingly influential role because of their polarity. The liquid crystal properties mainly the clearing temperature and enthalpy of transitions are influenced by the end chains. It is well known that physical and chemical properties of the liquid crystal molecules change with the length of alkyl chains [5].The spectroscopic and structural aspects pose big challenges to the interplay between theories, and experiments, with important implications to the photophysics, and chemistry of the materials containing electron- donating, and electron- withdrawing substituent effects [6]. In general, two types of structural modifications are possible in any chemical system: chain and ring. The present article involves the modification of first type. The Schiff’s base liquid crystals occupy a special place, among many chemical systems that are found to exhibit a liquid crystalline nature, as they present scenario with a wide incidence of polymorphism [7]. N (p-n-alkoxy benzylidene) p-n-alkyl aniline are popularly known as nO.m compounds represent an interesting case of liquid crystals owing to the presence of oxygen atom bridging the essential rigid core to the flexible end chains [8]. It is often found that the lower members of homologous series are purely or predominantly nematic, while the higher members, the smectic state encroaches on, and eventually prevents the observation of nematic. Though much of the experimental work has been carried out to infer the details of growth of nematic liquid crystal from isotropic liquid, the scarcely distributed data across the growth of smectic phases from the isotropic liquid are yet to be furnished [9].The compounds chosen for the present theoretical investigations NAPHE1, and NAPHE2. The structural and electrochemical properties such as Highest Occupied Molecular Orbital, HOMO (H), Lowest Unoccupied Molecular Orbital, LUMO (L) energies, and energy gap (Eg = EL - EH) have been investigated. Further, experimental investigations on the four synthesized compounds namely; N (p-n-Octyloxy benzylidene) p-n-Pentyl aniline (8O.5), N (p-n-Octyl benzylidene) p-n-Pentyloxy aniline (8.O5), N (p-n-Octyloxy benzylidene) p-n-Pentyloxy aniline (8O.O5) and N (p-n-Octyl benzylidene) p-n-Pentyl aniline (8.5) have been carried out. In the homologous series, the Octyl group acts as a boundary between nematic and smectic polymesomorphism. The position of oxygen atom present in the terminal chain varied from either side of the core, on both sides and is removed from the terminal chain. The observed values are discussed in the light of earlier data available on Schiff’s base nO.m compounds [10]. An examination of thermodynamic data reveals that NAPHE1, and NAPHE2 molecules exhibit nematic- isotropic transition at 380.2K, and 406.1K [11] respectively.
2. Theoretical and Experimental Details
2.1. Theoretical Details
- The first method is based on density functional theory (DFT). An understanding of absorption behavior requires the knowledge of molecular orbitals properties, spectral shifts, and appropriate excited states. Moreover, their photophysics, and chemistry represent a challenge in understanding of the excited states dynamics. The main difficulties against reliable theoretical approaches are concerned with the size of systems, and the presence of strong electron correlation effects. Both the properties are difficult to treat in the framework of the quantum mechanical methods rooted in the Hartree–Fock (HF) theory. The density functional theory (DFT) is successful to evaluate a variety of ground-state properties with accuracy close to that of the post-HF methods [12]. An important factor that determines the accuracy of time- dependent density functional theory (TDDFT) excitation energies is the exchange correlation functional used in the calculation. The use of these hybrid functionals yields good accounts of the vertical excitation energies of the excited states with substantial charge transfer character. In this context, the remarkable structural predictions have been obtained especially, using the ‘hybrid’ density functionals [13] such as Becke3-Lee-Yang-Parr (B3LYP) [14] combining ‘exact exchange’ with the gradient-corrected density functionals. As a consequence, there is currently a great interest in extending density functional theory (DFT) to excited electronic states. The time- dependent density functional theory (TDDFT) approach offers a rigorous route to the calculation of vertical electronic excitation energies, and the other spectral characteristics [15].
2.2. Experimental Details
- The compounds are prepared by condensation of the corresponding benzaldehyde (0.1mole) and aniline (0.1mole) on refluxing with absolute ethanol in the presence of few drops of glacial acetic acid [16]. After refluxing the reactants for four hours, solvent is removed by distillation under reduced pressure and the pure compound is recrystallized from absolute ethanol at low temperature. The optical textural observations [17] are made with an OLYMPUS DX 50 polarizing microscope equipped with DP 10 CCD display, in conjunction with an INSTEC temperature controller of accuracy ±0.1°C. The calorimetric investigations are carried out using a Perkin-Elmer DSC-7 instrument.
3. Results and Discussion
- The chemical structures of NAPHE1 and NAPHE2 have been shown in Figure 1. The density functional theory (DFT) optimized electronic structures have been shown in Figure 2. The general molecular structure of benzylidene aniline compounds is shown in Figure 3.
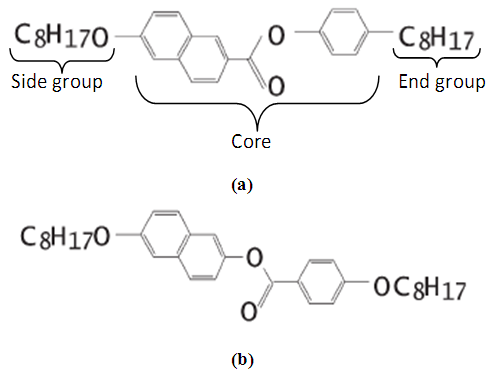 | Figure 1. The chemical structure of NAPHE1 (C33H44O3), and NAPHE2 (C33H44O4) molecules |
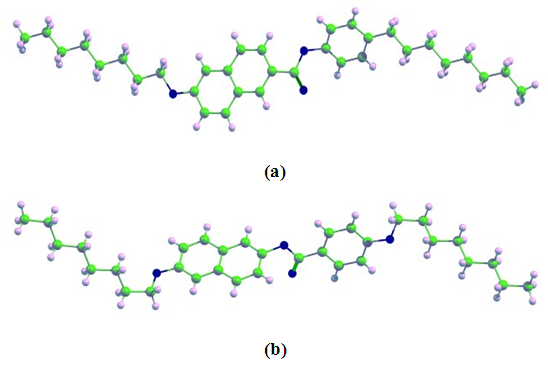 | Figure 2. The density functional theory (DFT) optimized electronic structures of (a) NAPHE1 and (b) NAPHE2 molecules (Green: Carbon, Blue: Oxygen, and Pink: Hydrogen atoms) |
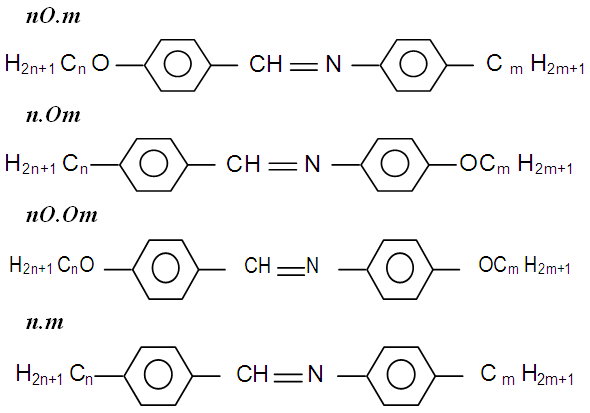 | Figure 3. The general structure of nO.m, n.Om, nO.Om and n.m compounds |
3.1. Part First: Theoretical
3.1.1. Substituent Induced Shifts
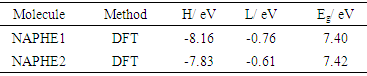
- The density functional theory (DFT) data shows that NAPHE1 molecule exhibits absorption maxima (λmax) at 201.76nm, and NAPHE2 at 239.84nm. Thus, the substitution of oxygen atom (with inversion of direction of ester linkage) in NAPHE1 molecule (forming NAPHE2) leads to a red-shift (the shift of absorption maxima to a longer wavelength), and also causes the hypochromic effect (decrement in absorbance). The HOMO (H), LUMO (L) energies, and energy band gap (Eg) values have reported in Table 1. Evidently, the HOMO (H), LUMO (L) energies, and energy gap (Eg) values have been found to be increased due to the substitution.
|
3.1.2. Intensity Profiles of NAPHE1 and NAPHE2
- The oscillator strength (f) is a dimensionless quantity that expresses the probability of absorption of electromagnetic radiation in transitions between energy levels of an atom or molecule. It indicates the allowedness of electronic transitions in a molecule, and it is particularly valuable as a method of comparing ‘transition strengths’ between different types of quantum mechanical systems. A graph has been plotted between wavelength, and oscillator strength (f) (Figure 4) to understand the intensity profiles of the compounds. It may be observed from the figure that NAPHE1 molecule exhibits the highest oscillator strength (f) at 257.50nm, while NAPHE2 molecule at 240.90nm. Further, NAPHE1, and NAPHE2 molecules exhibits the last intensity peak around 330nm, and 320nm respectively. This indicates the much flexibility of NAPHE1 molecule for electronic transitions over a long wavelength region. Further, this causes high photo sensitivity for NAPHE1 molecule, which may be exploited for electro-optic applications. The continuous decrease in oscillator strength (f) (Figure 4) clearly indicates the breakage of aromatic rings with respect to the higher wavelengths, and subsequently loosing the photo sensitivity.
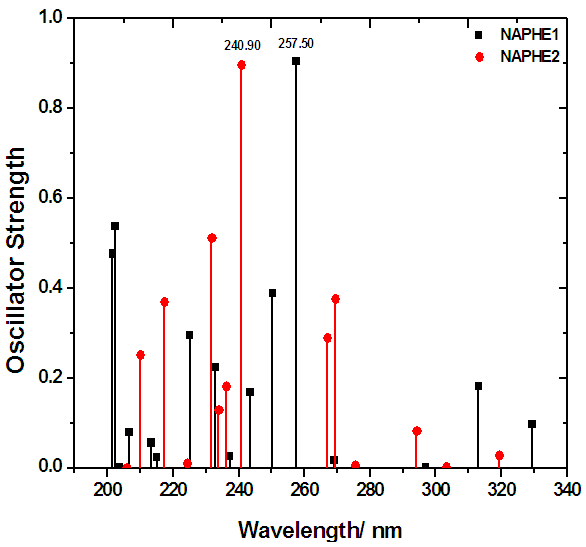 | Figure 4. Intensity profiles of NAPHE1 and NAPHE2 molecules using density functional theory (DFT) data |
3.2. Part Second: Experimental
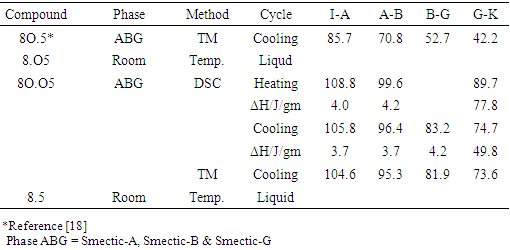
- The phase variants exhibited by synthesized compounds along with the phase transition temperatures and the corresponding enthalpy values are listed in Table 2. The phase transition temperature observed by thermal microscopy is found to be reasonable agreement with the DSC thermogram as shown in Figure 5.
|
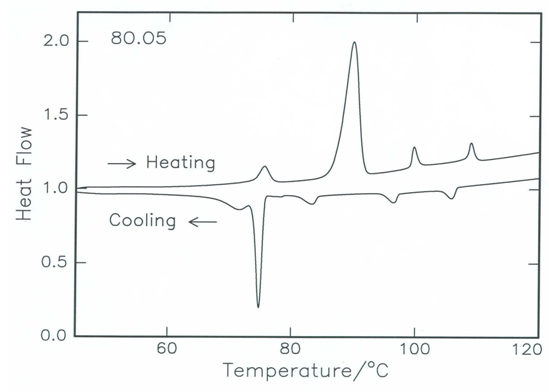 | Figure 5. The DSC heating and cooling thermogram of 8O.O5 compound at scan rate 10°C/min |
 | Plate 1. Smectic-A focal conic fan texture observed in 8O.O5 compound at 104.6°C |
 | Plate 2. Smectic-A to Smectic-B transient transition bars observed in 8O.O5 compound at 95.3°C |
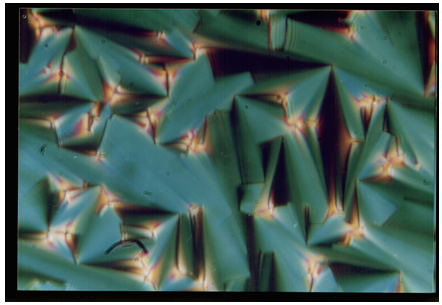 | Plate 3. Smectic-B texture observed in 8O. O5 compound |
 | Plate 4. Paramorphic Smectic-G textures observed in 8O. O5 compound at 81.9°C |
|
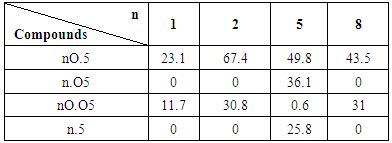
3.2.1. Influence of Oxygen’s Position on Compounds
- The position of oxygen in the aldehyde side, N (p-n-Octyloxybenzylidene) p-n-Pentyl aniline (8O.5) compound exhibiting its smectogenic character as smectic-A, smectic-B and smectic-G at above room temperature [18]. However, the change in position of oxygen from aldehyde side to the aniline side N (p-n-Octylbenzylidene) p-n-Pentyloxy aniline (8.O5) causes the compound to become room temperature liquid. But the effect is less prominence in the case of middle homologue (5O.5 and 5.O5) because both are exhibiting liquid crystalline nature at above room temperature level [21]. Furthermore, in lower homologous series like 1O.5, 1.O5 and 2O.5, 2.O5, there is no pronounced effect is observed on melting/clearing temperature, but major effect, quenching of liquid crystallinity is observed in case of 1.O5 and 2.O5 [22]. The change in position of oxygen has a pronounced effect on higher homologous series compared to lower homologous. The thermal stability of the compound 8.O5 is greatly affected and it leads to below room temperature level. This is due to the change in position of oxygen as well as contribution of alkyl chain length. It may be concluded from the above fact that the alkyl chain length is also actively participating in polymesomorphic property, melting and clearing temperature of the liquid crystalline compound.
3.2.2. Pronounced Effect of Oxygen
- The melting/clearing temperatures of N (p-n-alkoxybenzylidene) p-n-alkoxyaniline (nO.Om) compounds are very high, above 100°C in all cases of lower, middle and higher homologous series. An alkoxy group present in both sides of the rigid core is expected to enhance the stability by its oxygen unshared–pair overlap with the associated benzene ring [23]. In the case of N (p-n-alkylbenzylidene) p-n-alkylaniline (n. m), the absence of oxygen in rigid core causes the melting/clearing temperature to room temperature/below room temperature level. In both the cases, lower and higher homologous series (1.5, 2.5 and 8.5), the effect is merely same; the thermal stability of compound went to below room temperature. However, in middle homologue n = m = 5, 5.5 compound, the existence of liquid crystallinity is at and above the room temperature [21]. It is well known that physical and chemical properties of the liquid crystal molecules change with the length of alkyl chains. Subsequently, an identical alkyl chain length (5.5) present in both sides of the rigid core may responsible for the existence of liquid crystallinity, thermal stability and the change in length without oxygen has a pronounced effect.
4. Conclusions
- The salient features of the present work are as follows:1. NAPHE1 exhibits the last intensity peak at longer wavelength compared to NAPHE2. This shows that NAPHE1 is much flexible for electronic transitions over a long wavelength region. Further, this causes the intensity peaks over a large wavelength region, and high photo sensitivity for NAPHE1, which may be useful for electronic applications.2. The HOMO (H), LUMO (L) energies, and energy gap (Eg) have been found to be increased due to the substitution of oxygen. Further, it also causes the lower band gap in NAPHE1 compared to NAPHE2 molecule. Hence the conductivity is expected to be high for NAPHE1.3. The placement of oxygen atom plays an influential role in forecasting the polymesomorphism, and the alkyl chain length further tunes the occurrence of phase variant in benzylidene aniline compounds. 4. Although the major effects may be due to the presence/absence of oxygen in the terminal chain, a possible contribution from alkyl chain length could not be ruled out.5. The alkyl chains can be regarded as source of entropy to realize a given condensed state.
ACKNOWLEDGEMENTS
- One of the authors, Seema Prasad, is thankful to the UGC, New Delhi, India for providing financial support as JRF under the RGNF scheme.