Bahjat B. Kadhim, A. A. AL- Rubaiee, Methaq T. Matrood
Department of Physics, College of Science, Al-Mustansiriyah University, Baghdad, Iraq
Correspondence to: A. A. AL- Rubaiee, Department of Physics, College of Science, Al-Mustansiriyah University, Baghdad, Iraq.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Abstract
Nanotechnology is being applied to improve drug delivery in a number of ways. One of these ways is via built drug carriers. Nanosized particles of drug carriers are optimized for absorption of drugs through inhalation therapy. Modeling and simulation of nanocrystal parameters of the 5-Fluorouracil with indium gallium phosphide in diamantane structure have been performed with Gaussian 09 program. Density functional theory has been used for In5Ga2P7 nanocrystal, 5-Fluorouracil drug. Optimization plus frequency at the ground state level, PBEPBE, 3-21G basis sets has been investigated. The charges for all are equal to zero charges. Molecular orbital theory has been used to find highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies. Total energy, ionization potential and electron affinity have been calculated for In5Ga2P7 nanocrystal with 5-Fluorouracil drug.
Keywords:
Modeling, Simulation, Diamantane, Drug carrier, Density functional theory, and nanocrystal
Cite this paper: Bahjat B. Kadhim, A. A. AL- Rubaiee, Methaq T. Matrood, Structural and Electronic Properties of InGaP Nanocrystal Diamantane Drug Carrier, American Journal of Materials Science, Vol. 7 No. 1, 2017, pp. 12-17. doi: 10.5923/j.materials.20170701.02.
1. Introduction
The number of products based on new drug delivery systems has significantly increased in the past few years, and this growth is expected to continue in the near future. Recent advances in the field of genomics have accelerated research of biopharmaceuticals, and today a large number of companies are busy developing protein- and peptide-based drugs [1]. These biopharmaceuticals present challenges to drug delivery scientists because of their unique nature and difficulty in delivery through conventional routes [2]. Fluorouracil (5-FU) is medication drug used in cancer treatment. It is a suicide inhibitor and works through irreversible inhibition of thymidylate synthase. It belongs to the family of drugs called the antimetabolites [3]. It is on the World Health Organization's List of Essential Medicines, the most important medications needed in a basic health system [4]. Antimetabolite drugs work by inhibiting essential biosynthetic processes, or by being incorporated into macromolecules, such as DNA and RNA, and inhibiting their normal function. The fluoropyrimidine 5-fluorouracil (5-FU) does both [5].Nanoparticles are especially adaptable for delivery non – soluble and hydrophobic drugs. The size domain of particles between 10 and 100 nm lies between the size of materials that are extracted from the blood by microtubule filtration in the kidneys, and the sizes that are trapped by the liver, gall bladder, and liver, or that block capillaries in the lungs or other organs to cause emboli. If the dimensions of nanocrystal are smaller than twice the Bohr radius of the material it is made of, then quantum confinement occurs [6]. For the treatment of human diseases, nasal and pulmonary routes of drug delivery are gaining increasing importance [7]. The aim of this work is modeling and simulation to the creation and design of nanocrystal material of gallium indium phosphide which can be used for drug carrier to the place of destination in the human body using diamantane structure.
2. Materials and Methods
Geometry optimization is name for the procedure that attempt to find the configuration of minimum energy of the molecule [8]. The procedure calculates the wave function and the energy at starting geometry and then proceeds to search a new geometry of a lower energy [9]. This is repeated until the lowest energy geometry is found the procedure calculates the force on each atom by evaluating the gradient (first derivative) of the energy with respect to atomic positions sophisticated algorithms are then used at each step to select a new geometry [10].Diamondoids have been of great interest in recent years due to their role in nanotechnology, drug-delivery and medicine. The carbon-carbon framework of diamondoids constitutes the fundamental repeating unit in the diamond lattice structure. It is demonstrated that diamondoids are very stable compound [11, 12].The smaller diamondoid molecules, with the general chemical formula C4n+6H4n+12: adamantane (C10H16), diamantane (C14H20), and tiramantane (C18H24). Each of these three lower adamantologues has only one isomer [13].DFT is a computational quantum mechanical modeling method used in physics, chemistry and material science to investigate the electronic structure (principally the ground state) of many body systems, in particular atoms, molecules and condensed phases [14, 15]. With this theory the properties of a many electron system can be determined by using functional i.e. functions of another function which in this case is the spatially dependent electron density [16]. Hence the name density functional theory comes from the use of functional of the electron density [17].Indium Gallium phosphide (InGaP) is a semiconductor composed of indium, gallium and phosphorus. The InXGa1-XP ternary alloy is an attractive material for the preparation of variety of optoelectronic and microelectronic devices. The band gap energy is usually determined using a parabolic interpolation between (GaP) and (InP) [18].Theophylline is used to prevent and treat wheezing, shortness of breath, and chest tightness caused by asthma, chronic bronchitis, emphysema, and other lung diseases. It relaxes and opens air passages in the lungs, making it easier to breathe [19]. DFT partition the total energy as [20, 21]: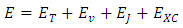 | (1) |
where 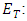 Electronic kinetic energy,
Electronic kinetic energy, 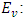 Electronuclear interaction energy,
Electronuclear interaction energy, 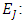 Electron-electron repulsion, and
Electron-electron repulsion, and 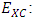 Exchange correlation term. According to Koopman's theorm in which the frontier orbital energies are given as [23]:
Exchange correlation term. According to Koopman's theorm in which the frontier orbital energies are given as [23]: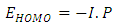 | (2) |
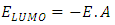 | (3) |
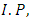 ionization potential and
ionization potential and 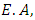 electron affinity.For the equilibrium system (e.g. atoms or molecules), let E(N) represent a ground state electronic energy as a function of the number of electrons (N). It is well-known the derivative of E(N) with respect to (N) at a constant external potential,
electron affinity.For the equilibrium system (e.g. atoms or molecules), let E(N) represent a ground state electronic energy as a function of the number of electrons (N). It is well-known the derivative of E(N) with respect to (N) at a constant external potential, 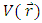 , the chemical potential
, the chemical potential 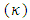 or the electronegativity
or the electronegativity 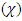 of the absolute negativity are [24]:
of the absolute negativity are [24]: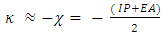 | (4) |
The theoretical definition of chemical hardness 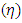 has been provided by the density functional theory as the second derivative of electronic energy with respect to the number of electrons N at a constant external potential
has been provided by the density functional theory as the second derivative of electronic energy with respect to the number of electrons N at a constant external potential 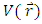 [25]
[25]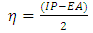 | (5) |
Equation (5) shows that chemical hardness is the resistance of the chemical potential to change in the number of electrons of the highest occupied and lowest unoccupied molecular orbital (HOMO and the LUMO energies) of the neutral molecule and is known as orbital-vertical [26, 27].All the computational studies were carried out using the density functional theory (DFT) implemented in the Gaussian 09W suite of programs. 27 Gaussian, a commercial quantum chemical software package from Gaussian incorporation is considered to be the industry standard in the area of molecular modeling and computational chemistry. Gaussian is capable of running all of the major methods in molecular modeling, including molecular mechanics; Ab-initio; semi empirical, and density functional theory (DFT). The molecular properties of the compounds have been computed by DFT using the standard 3-21G basis set. In the DFT calculations the Lee, Yang and Parr correlation functional is used together with Becke’s three parameters exchange functional B3LYP. Conformational analysis of the molecules has been performed to have an idea about the lowest energy structures of the species.
3. Results and Discussion
The optimization structure for In5Ga2P7 diamantane nanocrystal was calculated by using Gaussian 09 program. Figure (1) can be shown that the geometric structure of InGaP diamantane nanocrystal, 5Fluorouracil (C4H3FN2O2) and the number of atoms, so that these atoms in the molecule are numbered according to their order in the molecule specification section of the input. Figure (2) shows the optimized structure of 5-Fluorouraci-In5Ga2P7 Diamantane with PBEPBE/3-21G method.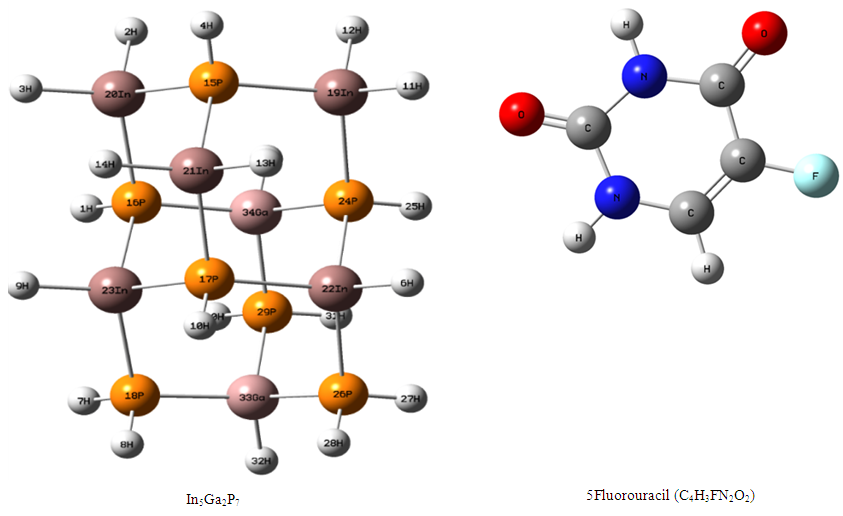 | Figure (1). The optimized structure of InGaP diamantane nanocrystal and 5Fluorouracil with PBEPBE/3-21 G method |
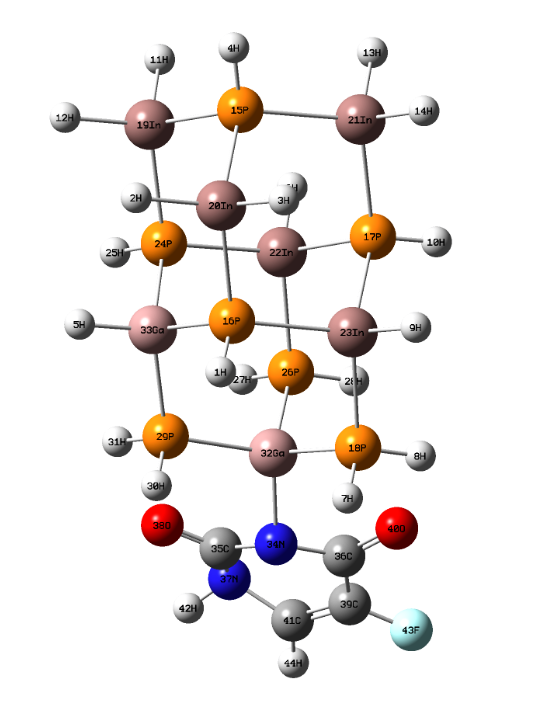 | Figure (2). Optimized structure of InGaP diamantane nanocrystal with PBEPBE/3-21 G method |
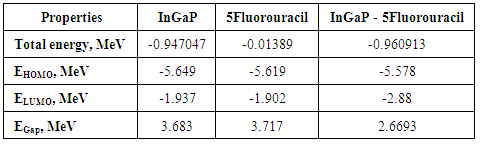
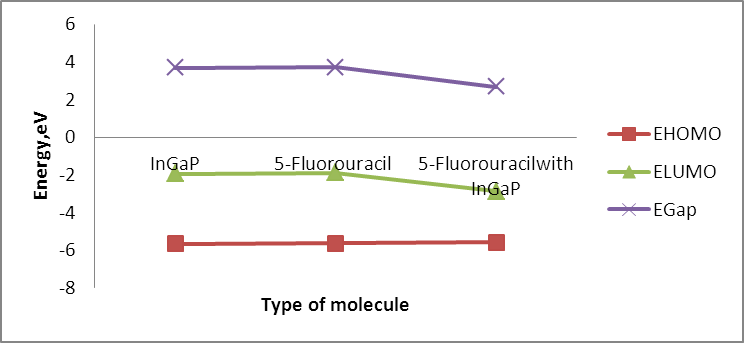
Table (1) represents the total energy for In5Ga2P7 diamantane, 5-fluorouracil drug and In5Ga2P7 diamantane binding 5-Fluorouracil. The total energy for In5Ga2P7 diamantane binding 5-Fluorouracil is less than the total energy for In5Ga2P7 diamantane, shown that the total energy decreases with increasing the number of Ga atoms using PBEPBE/3-21G (basis sets).Table (1). Total energy, HOMO, LUMO, and energy gap for InGaP nanocrystal and drug –InGaP. nanocrystal
 |
| |
|
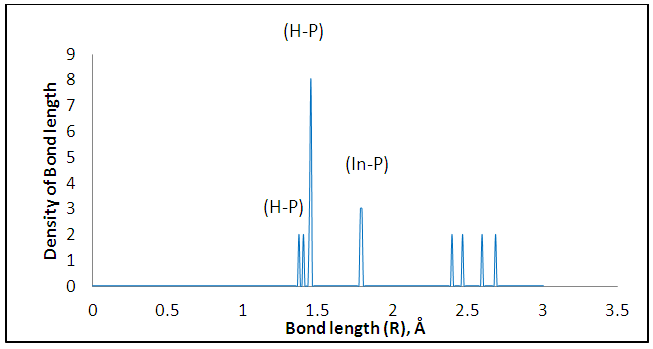
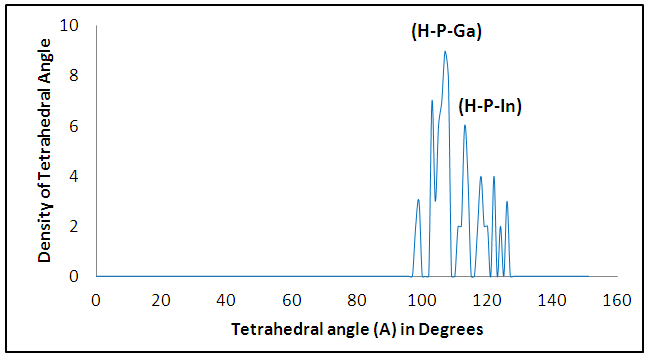
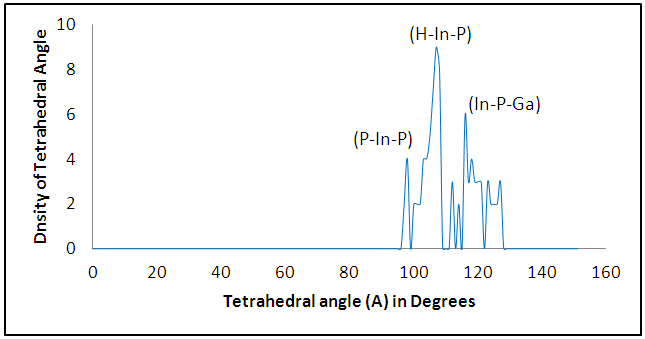
The size dependence of the energy is linear inversely proportional. Note that the total energy decreases with increasing the number of atoms in the diamantane structure, can be explained based on the principle of Heisenberg as the outer electrons of each atom in the system spend more time in region between atoms bound according Coulomb law, so it is more stable compound for drug delivery.It can be seen from Figure (3) that the most thick bond length lays at around 1.44 Å for (H-P) bond and alternate densities are at 1.78 Å for (H-In) bond, 2.62 Å for (H-G) bond. It can be seen from Figure (4) that the most thick bond length lays at around 1.44 Å for (H-P) bond and alternate densities are at 1.79 Å for (In-H) bond, 2.29 Å for (P-Ga) bond as that from Figure (3). Figure (5) can be seen In5Ga2P7 tetrahedral angle as (105.831) Å and In5Ga2P7 diamantane bonded with 5-Fluorouracil (107.036) Å (Figure 6). Electronegativity, hardness, softness index for InGaP diamantane nanocrystal, 5-Fluorouracil, and with InGaP nanocrystal using PBEPBE/3-21G energy-vertical method, are shown in Figures (7) and (8). The properties that are displayed in Figures (7) and (8) for each property is computed by employing the difference between the total energies of the neutral InGaP diamantane and the ions of InGaP diamantane.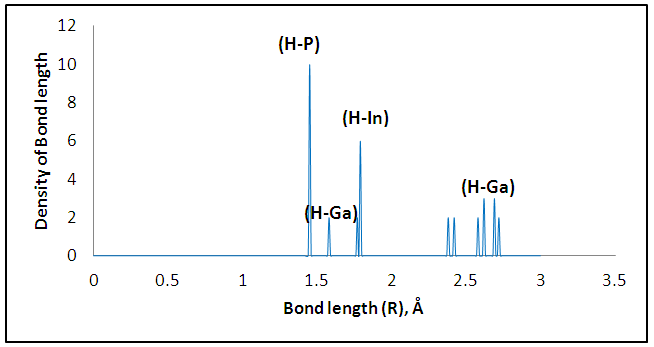 | Figure (3). Distribution of Bond lengths (R) in InGaP diamantane structure using PBEPBE/3-21G |
 | Figure (4). Distribution of Bond lengths (R) in InGaP diamantane with 5 Fluorouracil drug |
 | Figure (5). Distribution of tetrahedral angle (A) in degree in InGaP diamantane structure |
 | Figure (6). Influence of tetrahedral angle on Density of tetrahedral angle of 5-Fluorouracil-InGaP diamantane |
 | Figure (7). Effect of InGaP binding on the original 5-fluorouracil molecule with different electronic parameters |
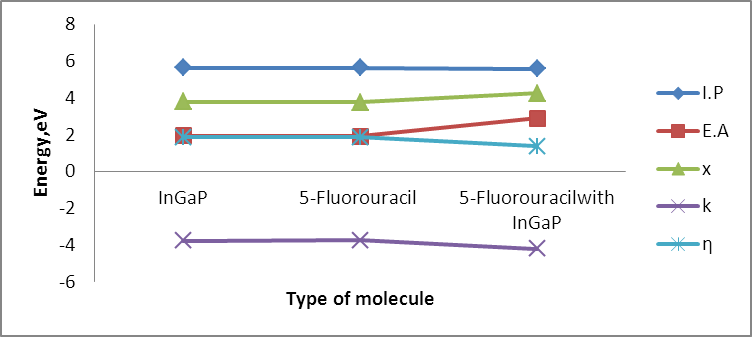 | Figure (8). Effect of InGaP binding on the original 5-Fluorouracil with different electronic parameter |
4. Conclusions
The geometry optimization using DFT with either exchange-correlation functional methods (PBEPBE/3-21G) for InGaP diamantane nanocrystal and 5furacil drug has been found in good and suitable to get the electronic properties. This study supplies a new data for 5Fluorouracil – InGaP diamantane nanocrystal for geometry optimization, total energy, and electronic states due to no previous studies for such type of nanocrystal structures.The total energies for 5Fluorouracil InGaP – diamantane nanocrystal causes decreasing in energy and more stable structure. The result indicates that the InGaP – diamantane working on the transfer drug without interacting with it. This means that the InGaP – diamantane is insulating material and this helps us in the binding process with the drug without the chemical reaction with any medication side effect.
References
| [1] | Ankith Kumar Reddy, Subhashis Debnath, and M. Niranjan Babu "A Review on Oral Control Drug Delevery System" International Journal of Pharmacy, (2013). |
| [2] | Wim H De Jong and Paul JA Borm "Drug Delivery and Nanoparticles: Applications and Hazards" International Journal of Nanomedicine (2008). |
| [3] | Ning Zhang, Ying Yin and Others "5-Fluorouracil: Mechanisms of Resistance and Reversal Strategies" Molecules 2008. |
| [4] | Thekra Kasimand and Mohammed T. Hussein "Study of the Electronic Structure of Indium Gallium Phosphide In0.5Ga0.5P Nanocrystals "Iraqi Journal of Physics, 2012. |
| [5] | Daniel B. Longley, D. Paul Harkin and Patrick G. Johnston “5-Fluorouracil: Mechanisms of Action and Clinical Strategies” 2003 Nature Publishing Group. |
| [6] | Brittany L. Oliva-Chatelain "Coating and Doping of Ge QDs" Rice University (2016). |
| [7] | Nidhi Mishra, Narayan Prasad Yadav and Vineet Kumar Rai " Efficient Hepatic Delivery of Drugs: Novel Strategies and Their Significance " Hindawi Publishing Corporation Biomed Research International (2013). |
| [8] | Mokhtaria Drissi, Nadia Benhalima and Youcef Megrouss "Theoretical and Experimental Electrostatic Potential around the m-Nitrophenol Molecule" Molecules 2015. |
| [9] | Ankan Das, Liton Majumdara and Sandip K. "Deuterium Enrichment of the Interstellar Medium" (2014). |
| [10] | FR. Jose .T.M “A theoretical study of the cation- pi interaction in alkali metal polyenes and polyene complexes” Thesis. Department of Chemistry, University of Calicut, (2007). |
| [11] | Hamid Ramezani, Mohammad Reza Saberi and G. Ali Mansoori "Diamondoids and DNA Nanotechnologies" (2007). |
| [12] | Gregory C. McIntosh, Mina Yoonand and Savas Berber "Diamond fragments as building blocks of functional nanostructures" Physical Review B 70, 045401 (2004). |
| [13] | A. Sliogeris, J. Tamuliene and R. Vaisnoras "Quantum Mechanical Investigations on Structure and Stability of the Smallest Diamondoids with Defects" Materials Physics and Mechanics 12 (2011) 186-194. |
| [14] | Vivek K. A and G. D. Agrawal "Organic Solar Cells: Principles, Mechanism and Recent Dvelopments" Volume: 03 Issue: 09 | Sep-2014. |
| [15] | Chong Chen "Ternary Alloy Material Prediction Using Genetic Algorithm and Cluster Expansion" Iowa State University 2015. |
| [16] | Klaus Capelle "A Bird’s-Eye View of Density-Functional Theory" 2006. |
| [17] | David S. Sholl and janice a. steckel "Density functional theory: a practical introduction" Wiley (2009). |
| [18] | Thekra Kasim and Abd Al-Raheem "Ab-Initio Study the Electronic Structure of InXGa1- XP Nanocrystals" (2013). |
| [19] | Peter J. Barnes and others "Theophylline" (2013). |
| [20] | M.-L. Man, C.-H. Lu, W.-K. Chen, Y. Li, and Y.-F. Zhang, "Theoretical and Computational Chemistry," Acta Phys Chim Sin, vol. 28, pp. 51-57, 2012. |
| [21] | Fouad N. Ajeel, Alaa M. Khudair and Anees A. Mohammed, "Density Functional Theory investigation of the Physical Properties of Dicyano Pyridazine Molecules", "International Journal of Science and Research", volume. 4, 4 issue, 1 Janary 2015. |
| [22] | Zeyad Adnan Saleh, Dukra Kamal Taha "Calculation of Ionization energies, electron affinities, hardnesses and electro negativites", using many bases set of many methods International Journal of Scientific & Engineering Research, Volume 5, Issue 12, December-2014. |
| [23] | Szabo, A.; Ostlund, N. S. Modern Quantum Chemistry: Introductionto Advanced Electronic Structure Theory; Dover Publications, Inc.: New York, 1989. |
| [24] | A. H. Raheem and others "Density Functional Theory Calculations of Thiophene - Phenylene Systems and Their Adducts" JOURNAL OF KUFA – PHYSICS Vol.5/ No.2 (2013). |
| [25] | Jenny Zevallos and Alejandro Toro-Labbé "A Theoretical Analysis of the Kohn-Sham and Hartree-Fock Orbitals and Their Use in the Determination of Electronic Properties" J. Chil. Chem. Soc. v.48 n.4 Concepción dic. 2003. |
| [26] | Ghaidaa. A. Hafedh Jaber "Study of the Effect of Cyano Subgroup on the Electronic Properties of Azulene Molecule: B3lyp-Dft Calculation" European Scientific Journal December 2013. |
| [27] | N. Surendra Babu “Computational Studies and Multivariate Analysis of Global and Local Reactivity Descriptors of Five Membered Heterocycles Molecules by Density Functional Theory (DFT)" International Journal of Pure and Applied Researches; Volume 1(1)/2015. |

 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML