-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Materials Science
p-ISSN: 2162-9382 e-ISSN: 2162-8424
2014; 4(4): 165-168
doi:10.5923/j.materials.20140404.02
A First Principle Study of Electronic Structure of In2ZnTiO6
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAshwani Kumar1, Durga P. Ojha2
1Department of Physics, National Defence Academy, Khadakwasla, Pune, 410231 MH, India
2P G Department of Physics, Sambalpur University, Jyoti Vihar, Burla, Sambalpur, 768019, India
Correspondence to: Ashwani Kumar, Department of Physics, National Defence Academy, Khadakwasla, Pune, 410231 MH, India.
| Email: |  |
Copyright © 2014 Scientific & Academic Publishing. All Rights Reserved.
The full potential Linearized augmented plane wave (FLAPW) method is employed to study the ground state and crystal properties of In2ZnTiO6. The electronic-energy band structure, site and angular-momentum decomposed density of states and charge-density contours of double perovskite In2ZnTiO6 are calculated by the FLAPW method with the generalized-gradient approximation using density-functional theory. From the analysis of density of states, we conclude that there is hybridization of Ti-d state with the O-p state, which implies that the interaction between the atoms of these two is highly covalent which is consistent with the calculation of electronic band structure as well as charge density studies. The equilibrium values, bulk modulus, and its pressure derivative have been estimated through optimization of the crystal structure of this material.
Keywords: Electronic band structure, Density of states, FLAPW
Cite this paper: Ashwani Kumar, Durga P. Ojha, A First Principle Study of Electronic Structure of In2ZnTiO6, American Journal of Materials Science, Vol. 4 No. 4, 2014, pp. 165-168. doi: 10.5923/j.materials.20140404.02.
Article Outline
1. Introduction
- Double perovskites with general formula A2B′B″O6 with A being an alkaline earth such as Ca, Ba, Sr or a lanthanide and B′, B″ are the transition metals are a matter of investigation since past decades [1-7]. Kang et al. [1] have studied the electronic structure of Ba2FeMoO6 using photoemission spectroscopy. Borges et al. [2] investigated the ferromagnetic metallic behaviour of Ca2FeMoO6, Sr2FeMoO6 and Ba2FeMoO6with curie temperature of 345 – 426 K. Moritomo et al. [3-4] shows a strong correlation between the curie temperature and the conductivity at room temperature which implies that the mobile conduction electrons mediate the exchange interaction between the local Fe3+ ions. Sarma [5] has pointed out the discrepancies between the manganites and the double perovskites and proposed the mechanism of occurrence of large TC in double perovskites Sr2FeMoO6. This mechanism has been extended to many other systems by Kanamori and Terakura [6-7]. In this model the hybridization of the Mo 4d (t2g) and Fe 3d (t2g) states plays the key role in stabilizing ferromagnetism at high curie temperatures [5-8] Based on band calculation using the Local Density approximation (LDA), Pickett [9] has proposed that the double perovskite La2MnVO6 can be a promising candidate for exhibiting half-metallic antiferromagnets (HM-AFM) characteristics. The electronic structure calculations were based on the assumption that both Mn and V ions are trivalent and more importantly a low spin state (t2g4eg0; S =1) resulted for all Mn3+ ions. Phillip et al. [10] studied the effect of structural changes and doping in the double perovskite A2CrWO6 (A = Sr, Ba, Ca) on the magnetotransport, the magnetic and optical properties together with band structure calculations. Phillip et al. have pointed out that the TC in double perovskites was discussed to depend sensitively on band structure and band filling in contrast to experimental results [11]. In double perovskites having structural formula A2B′B″O6, each transition metal site (B′B″) is surrounded by an oxygen octahedron and the A atoms site are situated in the holes produced by eight adjacent oxygen octahedral. The properties of double perovskite compounds are determined by the relative sizes, valencies and ordering of the A and B site ions.The study of elemental structures plays a pivotal role in solid-state physics [12]. The structural study gives insight into the properties of the elements. In the present paper, the electronic structure (density of states, band structure) of In2ZnTiO6 (IZT) are calculated by full potential linearized augmented plane wave (FLAPW) method [13] within the generalized gradient approximation (GGA) [14] as the material is a promising candidate for optoelectronic applications.
2. Computational Details
- The ground-state properties of IZT are investigated employing the FLAPW method within the generalized gradient approximation (GGA). The structural parameters of the system were optimized at 2000-k points inside the irreducible Brillouin zone for integration. The full geometrical optimization gives an in-plane lattice constant a = b = c = 8.0 and c/a = 1.0 of pure In2ZnTiO6. Accurate optimizations of crystal geometries have come to serve as a stringent test for electronic structure calculations. We use our optimized parameters for the study of electronic structure calculations taking muffin tin radius (RMT) as 2.0, 1.9, 1.8 and 1.6 for In, Zn, Ti and O respectively. The total energy has been minimized and calculated with respect to the volume. In our calculation, the crystal structure of IZT has the space group symmetry Fm-3m. The Birch-Murnaghan relation for equation of state (EOS) is used to get the static equilibrium volume V0 (=940.39) as well as the bulk modulus B0 (= 123.55 GPa) and its pressure derivative B (= 3.0873) at zero pressure.
3. Results and Discussion
- In figure 1, we have calculated and minimized the total energy with respect to the volume. In our calculation, the crystal structure of IZT has the space group symmetry Fm-3m. The Birch-Murnaghan relation for equation of state (EOS) is used to get the static equilibrium volume V0 (=940.39) as well as the bulk modulus B0 (= 123.55 GPa) and its pressure derivative B′ (= 3.0873) at zero pressure. The pressure derivative of bulk modulus at zero pressure B0′ is a parameter of great physical significance in high pressure physics. It is related to a few other important thermo physical properties (like phase transitions, interphase energy, adsorption energy etc.) [14].
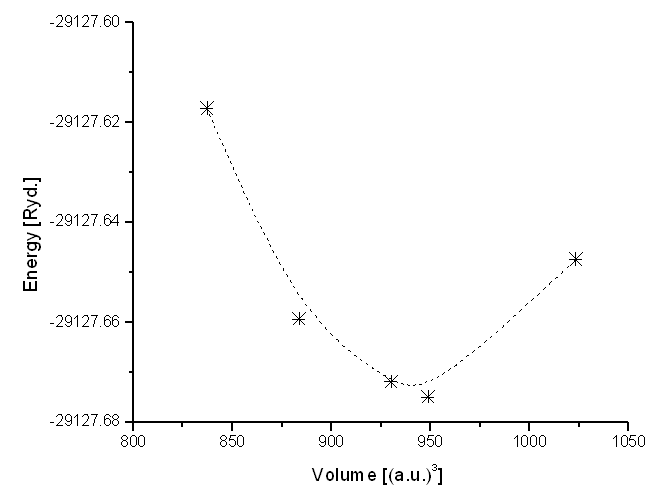 | Figure 1. Total Energy of In2ZnTiO6 as a function of cell volume |
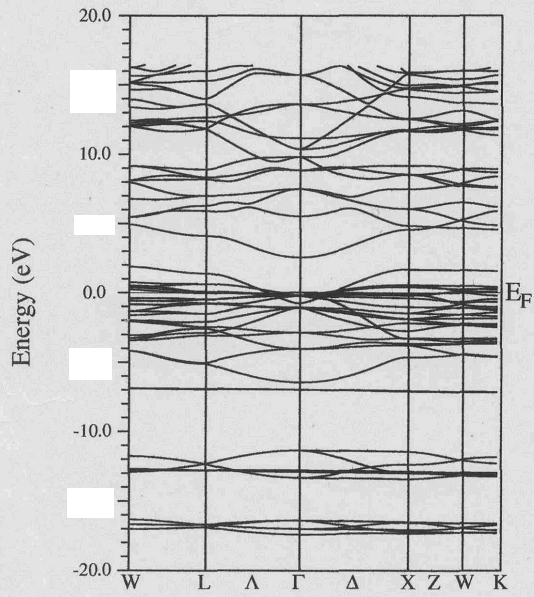 | Figure 2. The electronic band structure of In2ZnTiO6 along high symmetry directions |
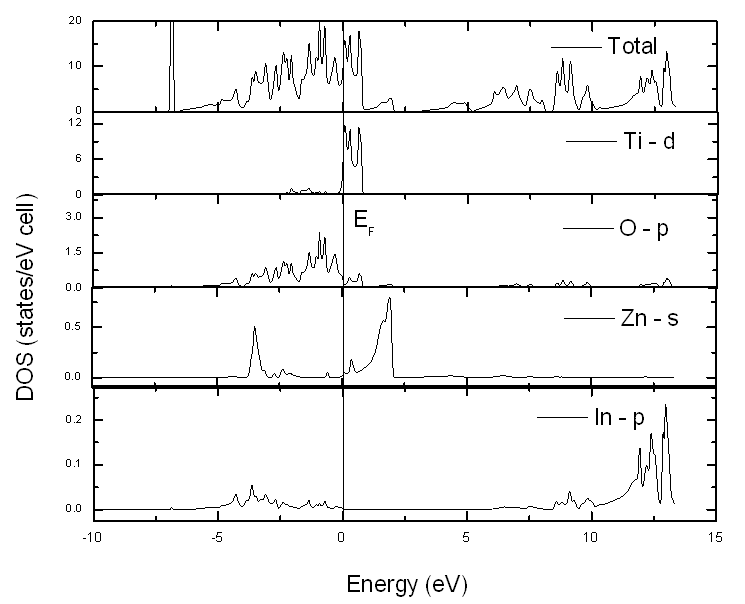 | Figure 3. Total DOS and PDOS around Fermi energy of In2ZnTiO6 calculated for the optimized lattice constant |
 | Figure 4. The electron density distribution of IZT in the (100) plane |
4. Conclusions
- We have performed and analyzed the FLAPW calculation of the double perovskite In2ZnTiO6 (IZT). The ground state and electronic structural properties have been obtained. From the angular momentum projected densities of states at 2000-k points inside the irreducible Brillouin zone for integration, it can be concluded that Ti-d state hybridizes with O-p states near Fermi level and the interaction between the atoms of these two is highly covalent which is further supported by the electron density distribution of In2ZnTiO6 in 100 plane.
ACKNOWLEDGEMENTS
- The financial support rendered by the DAE-BRNS, Mumbai, India is gratefully acknowledged.