Oleg Sannikov, Samuel Hanson, Paul Saunders, Kaitlin L. Branch, Nabyl Merbouh
Simon Fraser University, Department of Chemistry, 8888 University Drive, Burnaby, Canada
Correspondence to: Nabyl Merbouh, Simon Fraser University, Department of Chemistry, 8888 University Drive, Burnaby, Canada.
| Email: |  |
Copyright © 2019 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Abstract
Analysis of a simple NMR spectrum is fairly easy and straight forward for undergraduate students but analyzing a fairly complex NMR can be very daunting and requires a methodical approach. This work uses the ubiquitous Diels-Alder reaction of methylcyclopentadiene and maleic anhydride to introduce students to an NMR spectrum that consist of inseparable mixtures of isomers and their analysis using advanced NMR spectroscopic techniques. This laboratory experiment further outlines the use of derivatization techniques for the separation of isomeric mixtures by preparing diastereomers and using NMR spectroscopy for their identification.
Keywords:
Methylcyclopentadiene, Diels-Alder Reaction, Complex Mixtures, Nuclear Magnetic Resonance Analysis
Cite this paper: Oleg Sannikov, Samuel Hanson, Paul Saunders, Kaitlin L. Branch, Nabyl Merbouh, Introducing Complex NMR Mixtures at the Undergraduate Level: Analysis of the Diels-Alder Reaction between Methylcyclopentadiene and Maleic Anhydride (Part I), Journal of Laboratory Chemical Education, Vol. 7 No. 1, 2019, pp. 8-18. doi: 10.5923/j.jlce.20190701.02.
1. Introduction
In the past few decades, the ubiquity of the Diels-Alder reaction (DA) between cyclopentadiene (Cp) and maleic anhydride (MA) in the undergraduate curriculum left very little room for the improvement of this reaction. Although this reaction is well known, fully optimized and the products extensively described in the literature, [1-4], the astute student may pose the questions: What happens when the dienes or dienophiles are more complex? What happens when the resulting adducts cannot be separated? These are valid questions that this experimental work seeks to address. Since little has been done on this topic from a pedagogical point of view, these questions might be a good avenue/way to introduce undergraduate students to more challenging structure determination as well as diastereomer-separation techniques. We describe here an experiment that uses commercially available methylcyclopentadiene (MeCp) dimer. Students begin by analyzing, using NMR spectroscopy, the isomeric mixture resulting from its thermal cracking and then subsequently react the MeCp isomers with MA to obtain the corresponding DA adducts. The mixture of DA adducts is also analyzed by NMR spectroscopy to confirm the identity of the adducts present. The students then attempt to separate and characterize each of the isolated adduct.In this article, titled Part I, the students will investigate the formation of the DA adducts and fully characterize them assuming they are all endo isomers, while in Part II of this work, they will investigate the formation of the exo DA isomers under different conditions, fully characterize them and subsequently ascertain their exo or endo stereochemistries using nuclear Overhauser effect (NOE) NMR experiments and other necessary techniques.
2. Experimental
General. All reagents and solvents were purchased from Sigma-Aldrich and used without further purification. 1H-NMR spectra were recorded on Bruker Avance III 400 MHz spectrometer and were referenced to the residual chloroform (CDCl3) resonance at 7.26 ppm for proton and 77.16 ppm for carbon. All separations were performed using either chromatography on silica gel or with a Teledyne Combiflash system using ethyl acetate/hexanes mixture as eluent.Safety Precautions. Before performing the experiments, students should familiarize themselves with the hazards of the reagents and techniques used. The SDS for the compounds used should be consulted prior to the laboratory period, and appropriate safety measures should be taken. Care should be taken during the thermal cracking of the methylcyclopentadiene dimer due to the high temperatures required during the process and the complex setup. Students should also be aware of the exothermic reaction involved to produce adducts 1 and 2, and this experiment should be performed slowly and under appropriate cooling with ice. Safe operating procedures for the centrifuge used to vortex the reaction should be provided and followed. During the purification steps, volatile solvents should be handled safely in a fume hood or well-ventilated area.
2.1. Thermal Cracking of the Methylcyclopentadiene Dimer
A distillation apparatus consisting of a pressure equalizing dropping funnel, a 2-neck flask (100 mL), a Vigreux column fitted with thermometer adaptor and a thermometer, and connected to a water-cooled, jacketed condenser equipped with a receiver flask were used for this procedure, Figure 1.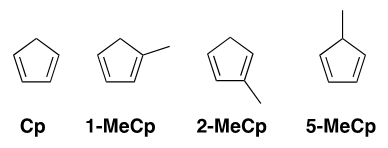 | Figure 1. MeCp dimer cracking set-up |
In the 2-neck flask, paraffin oil (50 mL) was first heated to approximately 270°C with stirring using a hotplate equipped with an aluminum block. Using the dropping funnel, the MeCp dimer was added to the hot paraffin oil in small portions (0.5 mL). This controlled dimer addition allowed the temperature on the top of the Vigreux column to be maintained between 65~72°C. The first ~ 2 mL of the distillate were discarded and the monomer was subsequently collected in a clean flask, cooled in an ice bath. The monomer was collected as a mixture of isomers. 1-MeCp and 2-MeCp were the predominant isomers as confirmed by 1H-NMR spectroscopy (Over 6 trials).
2.2. Diels-Alder Reaction of Methylcyclopentadiene Isomers with Maleic Anhydride
Finely grounded maleic anhydride (MA) (1.0 g, 10 mmol) was placed in a centrifuge tube fitted with a cap (15 mL) and cooled in an ice bath. Freshly cracked MeCp (2 ml) was slowly and carefully added into the centrifuge tube. The reaction is highly exothermic so the use of an ice bath is essential, and quantities of starting material can be reduced as well, as long as the ratio MA/MeCp is kept at 1:2 (m/v). The mixture is sporadically vortexed to help dissolve the unreacted MA, and once the MA is totally dissolved the reaction is left on ice for an additional 30 minutes. The purification of the Diels-Alder adducts was performed by adding pentane (3 × 5 mL) to the centrifuge tube and vortexing the reaction mixture for 30 seconds to dissolve the unreacted MeCp and removing the pentane layer manually using a Pasteur pipette (the Diels-Alder adducts and the pentane layers separated perfectly). The final product was dried using a flow of nitrogen to remove the last traces of pentane. In all cases, the reaction was reproducible, with yields averaging over 80% based on the MA used. [The reaction was performed using a ratio MA/MeCp (m/v) of 1 g / 1.5 mL, 1 g / 2 mL and 1 g / 3 mL and all gave similar yields (1.5 ~1.55 g). However the reaction mixture using a 1/2 ratio was easiest to purify, while the reaction with a ratio of 1/3 resulted in a waste of MeCp as the yield was not improved].
2.3. Diels-Alder endo Adducts Reaction with L-alanine Methyl Ester
A mixture of the Diels-Alder adducts (1.70 g, 9.55 mmol), triethylamine (1.20 g, 11.88 mmol) and L-alanine methyl ester hydrochloride (1.40 g, 10 mmol) in toluene (40 ml) was heated at reflux overnight. The resulting mixture was filtered, and the filtrate washed successively with 2M hydrochloric acid (2 × 30 mL), saturated aqueous sodium bicarbonate (2 × 30 mL), and brine (30 mL). The organic layer was collected, dried with MgSO4 and the solvent evaporated in vacuo. The resulting oil (1.83 g, 73% crude yield) was purified using column chromatography on silica gel or a Combiflash automated system.
2.4. L-alanine Methyl Ester Adduct Purification
The crude reaction mixture extract (0.5 g) was purified using flash liquid chromatography on a Teledyne CombiFlash Rf system equipped with a 24 g normal-phase RediSep Rf Gold silica column. The elution was done with an ACS grade hexanes/ethyl acetate mixture. The sample was dry packed using a minimal amount of Fisher 230-400 mesh normal-phase silica gel.The separation was done using the following parameters: Flow Rate –35 ml/min; Equilibration Volume –5.0 CV (Column Volume); Run length –20 CV; Fraction volume –7 mL; UV wavelength –254 nm & 280 nm. Gradient: 10% ethyl acetate – 50% over 13.1 CV; 50% ethyl acetate over 2.6 CV; 100% ethyl acetate over 4.4 CV. Diastereomer 1-MeCP-DA (80 mg) eluted first followed by 2-MeCP-DA (90 mg).This separation was performed under identical conditions on a crude mixture amount ranging from 0.5 g to 0.8 g with separation yields ranging from 10% to 40% for both adducts. This variation was attributed to the amount of silica gel used to dry pack the crude material.
3. Results and Discussion
3.1. Methylcyclopentadiene Dimer Thermal Cracking
The thermal cracking of the commercially available methylcyclopentadiene dimer typically results in a mixture of three isomeric products, Figure 2. While the resulting mixture has been identified to include traces of cyclopentadiene (Cp), 1-methylcyclopentadiene (1-MeCp), 2-methylcyclopentadiene (2-MeCp) and 5-methylcyclopentadiene (5-MeCp); [5-7] without prior knowledge of possible couplings in cyclopentadiene and its derivatives, the analysis and identification of the products can be challenging at the undergraduate level. Using a combination of double resonance experiments and theoretical calculations, the chemical shifts and coupling constants of the protons in 1-MeCp and 2-MeCp have been determined, albeit with some unresolved multiplicity patterns. [7] Another added complexity to the spectra of the mixture is the fact that 5-MeCp often appears as a trace and only rarely as an important fraction during the thermal cracking of the MeCp dimer. | Figure 2. Possible cyclopentadiene derivatives observed during the thermal cracking of the methylcyclopentadiene dimer |
Our more recent approach to address the complexity of this mixture was to introduce the students to a full package of NMR spectra comprising a proton (1H), a carbon (13C), an attached proton test (APT), a homonuclear correlation spectroscopy (COSY), a heteronuclear single-quantum correlation spectroscopy (HSQC) and a heteronuclear multiple-bond correlation spectroscopy (HMBC) NMR spectra to help with easier structural assignments. [8-10]Using all the spectra available, the structural assignment was made easier and no double resonance experiments or theoretical calculations were needed. A look at the 1H-NMR spectrum of the mixture shown in Figure 3 revealed the presence of proton signals in three different regions: two signals between 2.00 and 2.10 ppm, two signals between 2.86 and 3.00 ppm and four signals between 6.00 and 6.45 ppm. Based on the three possible structures of MeCp and the 13C information provided by the APT, Figure 4, in combination with the HSQC spectra, the methyl (CH3) peaks appear upfield (15.17 and 16.01 ppm) relative to methylene (CH2) peaks (41.44 and 44.89 ppm). It can be inferred with some degree of certainty that the peaks at 2.00-2.10 and 2.86-3.00 ppm belong to the methyl (-CH3) and methylene (-CH2-) protons, of MeCp respectively, while the peaks around 6.00-6.45 are those of the vinylic protons. Additionally, the APT showed the presence of two methyl and two methylene peaks only, but more importantly, no methine group (>CH-) between 0 and 50 ppm, hinting that only 1-MeCp and 2-MeCp isomers were present, Figure 4. (It is possible that 5-MeCp and Cp could still be present in the mixture in trace amounts below the detection limits of the instrument.)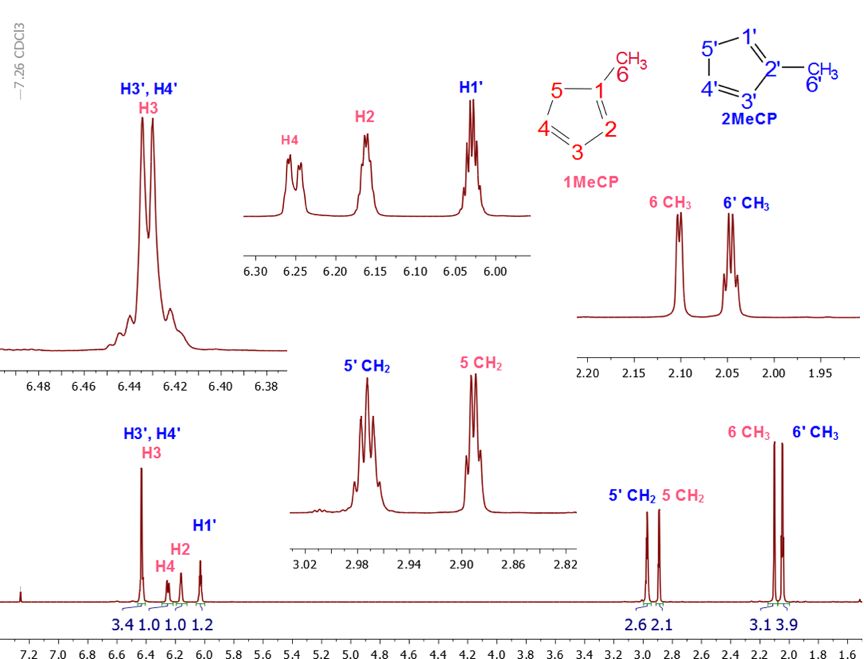 | Figure 3. 1H NMR of the mixture of methylcyclopentadiene isomers, zoomed in on the areas of interest |
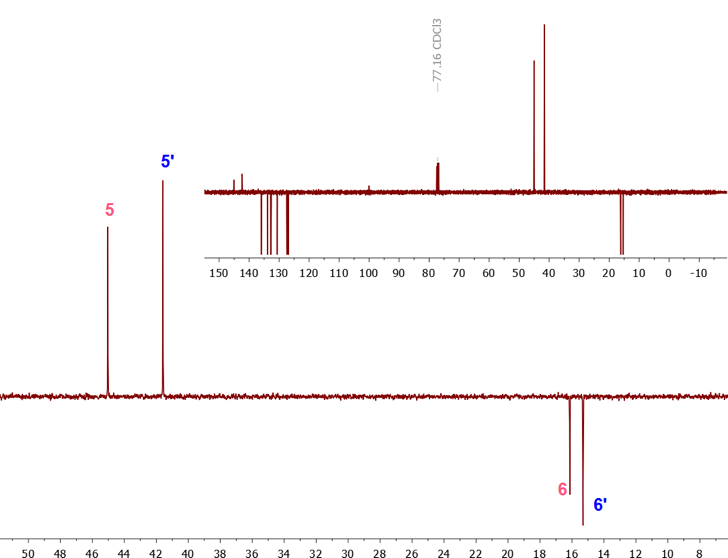 | Figure 4. APT spectrum (0 to 50 ppm region) of a mixture of methylcyclopentadiene isomers, with complete APT spectrum shown in indent |
The 1H-NMR spectrum showed a quartet and a doublet at 2.04 (J = 2 Hz) ppm and 2.09 ppm (J = 1.5 Hz), each accounting for 3.76 and 3 protons (methyl peaks) respectively, implying that the MeCp isomers are present in different proportions. As confirmed from integration, the methylene signals: a quartet- and a quintet-looking signals at 2.88 ppm and 2.97 ppm, corresponds to 2.01 and 2.48 protons, respectively.The integral values of the methyl and methylene signals can be used to establish the ratio of each isomer in the mixture using equation 1. The integral values reveal that 1-MeCp and 2-MeCp are present in a ratio of 44.4:55.6%, respectively. Similar values were reported by Ford [11] and McLean. [12] (Although the methyl resonances were used to calculate the proportion of each isomer in the mixture, using the methylene signals give identical results) | (1) |
Unfortunately, at the undergraduate level, the analyses of the coupling patterns and the peak shapes in the MeCp isomers are not trivial due to long range couplings as well as some possible second order effects. However, the assignments of all peaks could be readily accomplished using 2D-NMR experiments with the methyl and the methylene groups’ chemical shifts as starting points.To better understand the coupling pattern of the methyl protons in 1-MeCp, it should be noted that although four bonds’ couplings (4JHH) in saturated alkanes are rare, they are commonly observed in alkenes. Such couplings are pronounced in cyclopentadiene and its derivatives. Additionally, the cyclopentadiene ring shows several coupling constants of the same magnitude between the ring protons. [13]The methyl signal (H-6) of 1-MeCp at δ = 2.09 ppm appeared as a doublet due to four-bond allylic coupling (4J6-2 = 1.5 Hz) with the vinylic proton (H-2) at 6.14 ppm. This is supported by the COSY spectrum, Figure 5, which showed a correlation of the H-6 protons with the H-2 proton, but not with the methylene protons (H-5). The methylene proton of 1-MeCp, however, showed allylic coupling with the alkene protons H-2 (δ 6.14 ppm), H-3 (δ 6.24 ppm) and H-4 (δ 6.42 ppm) to give a quartet with coupling constants around 2 Hz.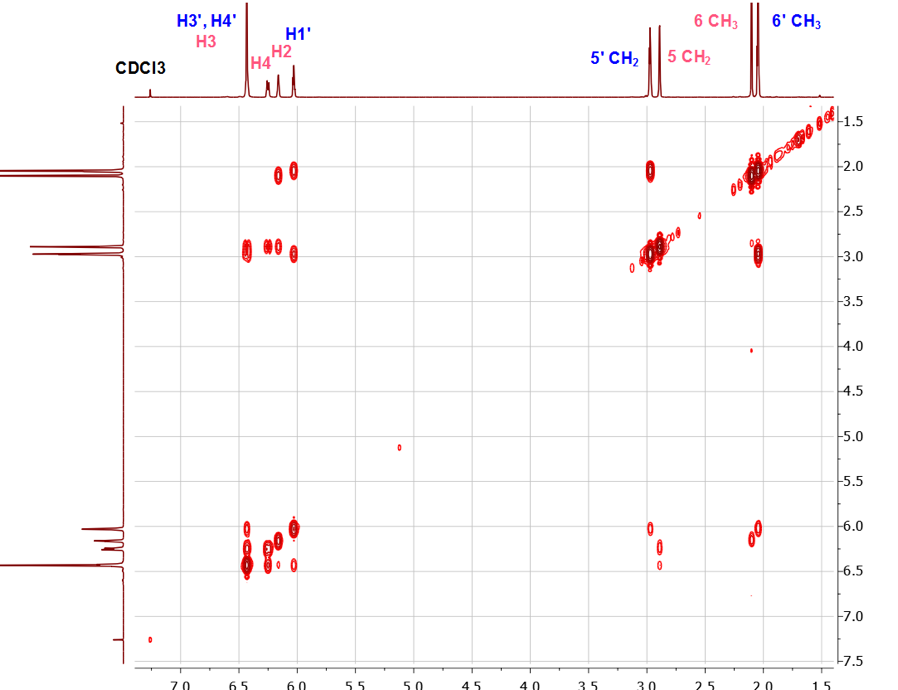 | Figure 5. COSY spectrum of the MeCp isomeric mixture |
The H-2 signal at 6.14 ppm appeared as a sextet because of couplings with the CH3 protons and the two vinylic protons H-3 and H-4. The H-4 signal, on the other hand, appeared at 6.24 ppm as a doublet of quartets due to the stronger coupling with H-3 (J = 5.4 Hz) and couplings with the two methylene (J = 1.5 Hz) protons and H-2 (J = 1.5 Hz). The chemical shift for the H-3 proton at 6.42 ppm coincides with the H-3 and H-4 protons of 2-MeCp isomer, thereby rendering the analysis of its multiplicity pattern impossible.In summary, the proton signals for 1-MeCp include a doublet at 2.09 ppm, a quartet at 2.88 ppm, a sextet at 6.15 ppm, a doublet of quartets at 6.27 ppm and a multiplet around 6.38-6.45 ppm. These signals correspond to the methyl, methylene, H-2, H-4 and H-3 protons, respectively.Similarly, the methyl signal of 2-MeCp at 2.04 ppm appeared as a quartet due to couplings with H-1, H-3 and H-4. H-1 appeared as a sextet due to couplings with the methyl protons and the other two vinylic protons (H-3 and H-4), which resonate at the same frequency at 6.42 ppm. H-3 and H-4 appeared as a doublet with a coupling constant of 5.4 Hz. The proton chemical shifts of 2-MeCp therefore include a quartet at 2.04 ppm (J = 2.0 Hz), a quintet at 2.97 ppm, a sextet at 6.02 ppm, a doublet around 6.42 ppm (J = 5.4 Hz) corresponding to the methyl, methylene, H-1, H-3 and H-4 protons, respectively. [2,3]When the students relied on the methyl groups’ assignments to start their investigation, it was useful to take a quick look at the HMBC spectrum at some point, Figure 6. Indeed, a quick prediction of the possible correlations for the methyl protons (H-6) allowed for an additional proof of their assignments. As seen in Figure 6, a correlation between H-6 of 1-MeCp was observed while the same correlation was not observed in the case of 2-MeCp.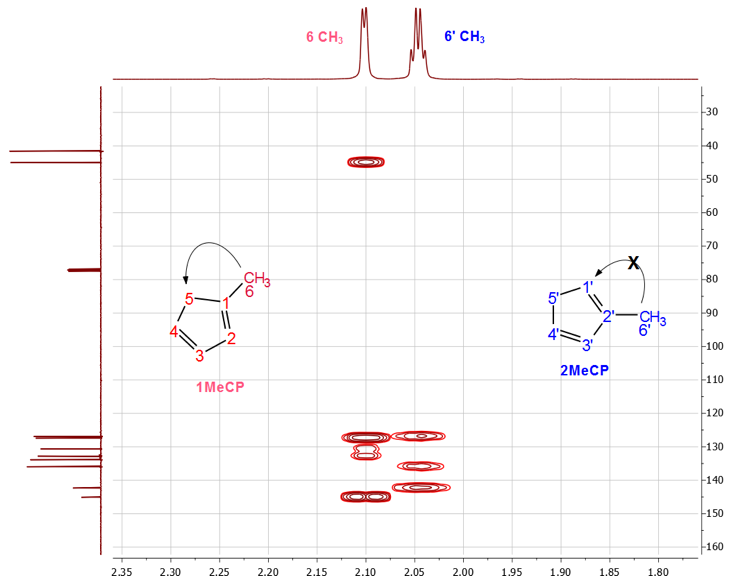 | Figure 6. HMBC spectrum of the mixture of 1 and 2. Correlation observed in 1-MeCp is not observed in 2-MeCp |
Once the MeCp isomer assignments are completed, the students can react the MeCp mixture with maleic anhydride (MA) to form the Diels-Alder Adducts under kinetic control (i.e. 0°C). [14] To avoid the use of any solvents that would need removal at the end of the reaction, MeCp could be used in slight excess as it was found to be a good medium for the reaction.
3.2. Products Formed from the Reaction of Methylcyclopentadienes with Maleic Anhydride
The MeCp mixture obtained from the cracking of methylcyclopentadiene dimer was reacted with maleic anhydride to give isomeric mixtures of methyl-5,6-dicarboxy-2-norbornene adducts. Theoretically, at least six products can be expected from such reaction (Eight if we take in account the possible presence of Cp in the mixture). However, based on NMR spectroscopy, fewer isomers were observed, which is not unexpected since the starting MeCp mixture contained mainly 1-MeCp and 2-MeCp and the reaction was performed under conditions of kinetic control (i.e. low temperature). The endo isomers of 1, 2 can be expected from such a reaction due to the kinetic conditions used for the synthesis, Figure 7. [15-16] (Note that the enantiomers of each of 1 and 2 will also be present.)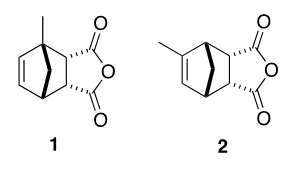 | Figure 7. Kinetic Diels-Alder adducts resulting from the reaction between the methylcyclopentadiene isomers and maleic anhydride |
A quick glance at the proton and carbon NMR spectra, Figure 8 and 9, showed the presence of very limited amounts of alkene protons (three) as well as methyl groups (two), further suggesting that the Diels-Alder adducts formed and their percentage composition could readily be identified.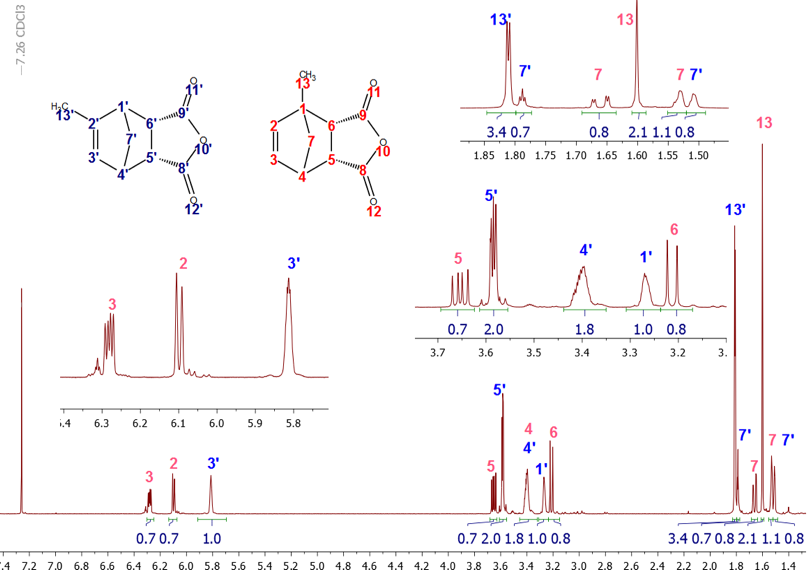 | Figure 8. 1H-NMR spectrum of the crude Diels-Alder reaction mixture |
 | Figure 9. APT-NMR spectrum of the crude DA reaction mixture |
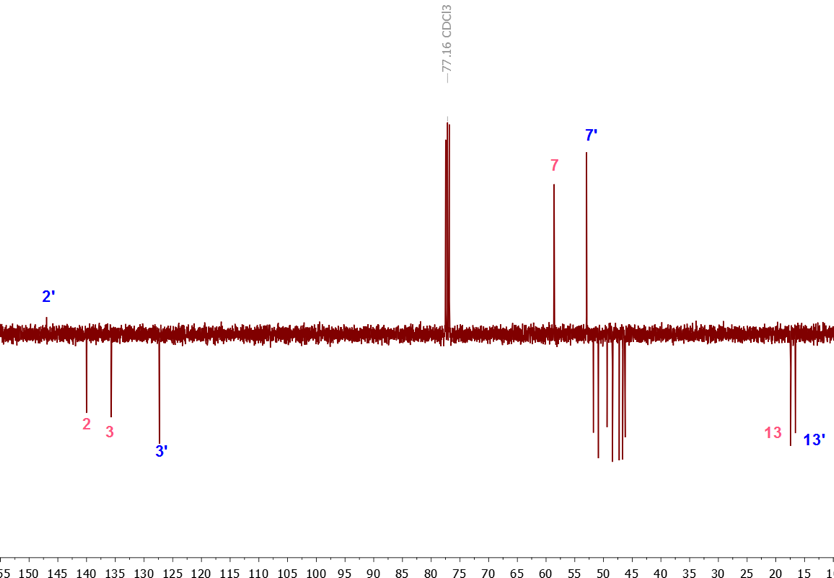
The 13C-NMR and APT spectra showed clearly the presence of two methyl groups only (16.5 and 17.3 ppm), confirming the presence of only two main adducts in the mixture. Additionally, the presence of 4 alkene carbons (127.2, 135.5, 139.9 and 146.8 ppm) one of which is quaternary (146.8 ppm) corroborating the presence of only two adducts, with one adduct having a tri-substituted double bond and one with a di-substituted double bond.The assignment of the alkene carbons and protons could subsequently be easily done using the COSY and the HSQC spectra. Indeed, the COSY spectrum shows clear coupling between the doublet of doublet at 6.31 ppm (J = 5.6, 3.0 Hz) and the doublet at 6.12 ppm (J = 5.7 Hz), while the multiplet at 5.84 ppm shows no coupling in the alkene region, Figure 10. The HSQC spectrum permits the rapid assignment of the carbon signals at 127.2 and 146.8 ppm to the isomer with the tri-substituted alkene (2) and the carbon signals at 135.5 and 139.9 ppm to the isomer with the di-substituted alkene (1).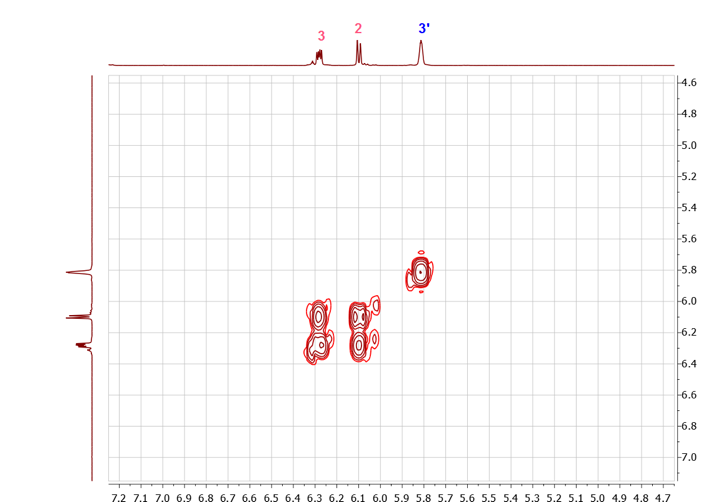 | Figure 10. Truncated COSY spectrum (5 - 7 ppm) of a mixture of methyl-5,6-dicarboxy-2-norbornene isomers, showing correlations of the methine protons and vinylic protons of adduct I |
With the alkene protons assigned to the two adducts present, the choice can be made to either continue using these alkene protons and the COSY spectrum to assign the rest of the NMR peaks or use the HSQC spectrum to assign the diastereotopic protons (H-7 and H-7’) present in the adducts. Either way, the HSQC spectrum can be used to assign the carbon and proton shifts of C-7 and H-7 and H-7’ to be 52.8 and 58.4 for C-7 and C-7’, respectively the pairs of H-7 and H-7’ protons to be 1.55 and 1.82 ppm for the first pair and 1.55 and 1.68 ppm for the second pair, albeit with a slight ambiguity in the assignment of the second H-7 proton. The HSQC spectrum also allowed the quick assignment of the methyl protons to be 1.63 ppm as a singlet and 1.84 ppm as a doublet (J = 1.7 Hz). Inspection of the COSY spectrum showed a coupling between the methyl peak at 1.84 ppm and the alkene proton at 5.84 ppm, further substantiating the 4J coupling constant of 1.7 Hz. The COSY spectrum readily assigns H-4 chemical shift to be 3.42 ppm in both adducts.At this point, it is important to remind the students that NMR spectra can help solve the structures using either the chemical shifts, the multiplicities, or the integrations of the peaks. Indeed, students can get lost in the complexity of the spectra and forget small details such as the integration of the peaks. Integrations in the 1H-NMR spectrum clearly show a ratio of 1 to 1.3 between adducts 1 (43 %) and 2 (57 %). This result is not unexpected given that the starting materials 1-MeCp and 2-MeCp were originally present in a similar ratio.Inspection of the integrations in the 1H-NMR spectrum helped assign the last peaks in the spectrum. Indeed, the doublet at 3.24 ppm (J = 8.1 Hz, 1H) and the doublet of doublets at 3.68 ppm (J = 8.1, 4.8 Hz, 1H) showed a strong correlation to each other and to H-4, and can be assigned to H-6 and H-5, respectively, in adduct 1. Using the COSY spectrum, Figure 11, the remaining two multiplets at 3.29 ppm and 3.60 ppm in the 1H-NMR spectrum could now be assigned to adduct 2, with the multiplet at 3.29 ppm assigned to H-1 and that at 3.60 ppm to H-4 and H-5 (overlapping signals).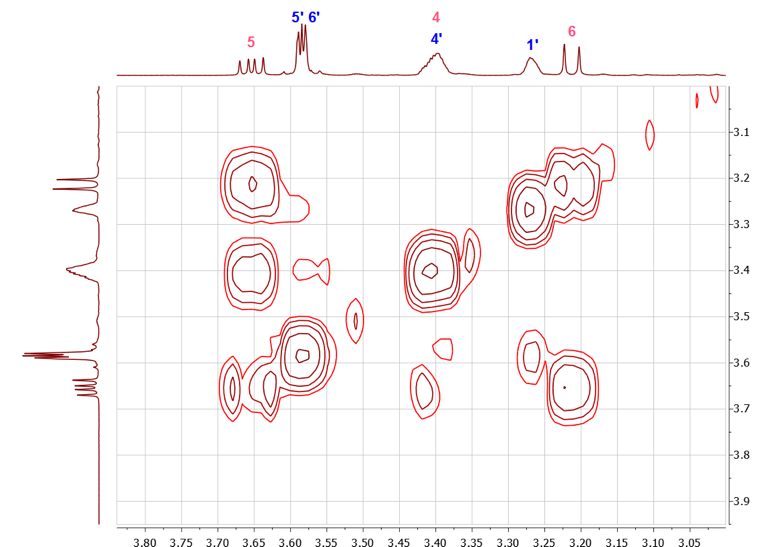 | Figure 11. Truncated COSY spectrum (3 - 4 ppm) of a mixture of methyl-5,6-dicarboxy-2-norbornene isomers |
3.3. Separation and Confirmation of the Structure of the Products Formed from the Reaction of Methylcyclopentadienes Mixture with Maleic Anhydride
As described throughout the literature, the isomers formed from the reaction of MeCp and MA are hard to separate and the characterization of the products is often done using the spectroscopic data of the isomeric mixture. [15-16]While such problem is not uncommon in organic chemistry, students have a plethora of techniques available to try solving this problem. The available techniques range from fractional distillation and recrystallization to the use of chiral salts’ derivatives and the synthesis of diastereomeric mixtures. The use of the diastereomeric mixture was investigated as it could be used on small material quantities, as well as oils, in the time undergraduate laboratories permit. (Furthermore such technique could be applied to other ubiquitous undergraduate experiments such as the reduction of ketones and the separation of their resulting isomeric mixtures)Interestingly, to unequivocally establish all NMR assignments discussed above and to separate isomers 1 and 2, a simple conversion of the DA adducts to norbornyl amino acid esters (3 and 4) proved to be a successful strategy. [17-18] The reaction of the DA adducts mixture with L-alanine methyl ester yielded new derivatives that could be readily separated by chromatography on silica gel (Either column or automated Combiflash). These new compounds (3a, 3b, 4a and 4b), shown in Figure 12, exhibited interesting NMR spectra and can be used to introduce the students to another facet of diastereomer separation.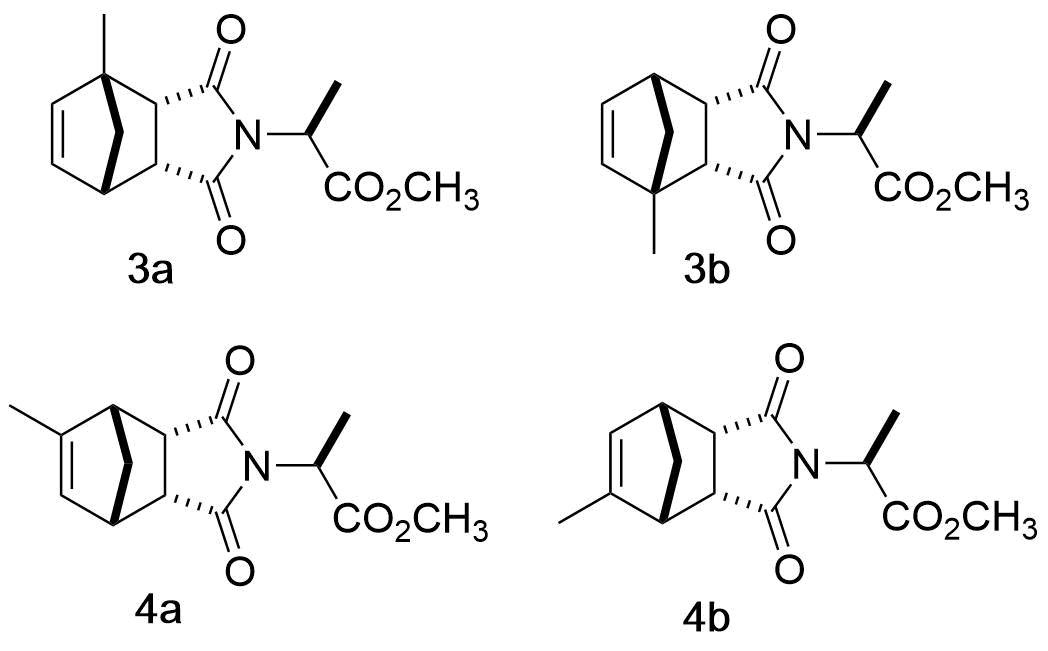 | Figure 12. Products of the reaction of the DA adducts with L-alanine methyl ester |
While the 1H-NMR spectra of the separated diastereomers (3a, 3b, 4a and 4b) could readily be interpreted due to differences in double bond substitution (Figure 13), the 13C-NMR spectra shown in Figure 14, showed duplication of each peak for both compounds, requiring the students to take a more careful look at the methylnorbornene derivatization. Such signal doubling should lead students to the conclusion that four compounds were formed in the Diels-Alder reaction between methylcyclopentadiene and maleic anhydride, namely two sets of enantiomers. That is, methyl-5,6-dicarboxy-2-norbornene adducts 1 and 2 each exists as a racemate. Reaction of each set of racemates with L-alanine methyl ester then yields four diastereomers, one for each enantiomer.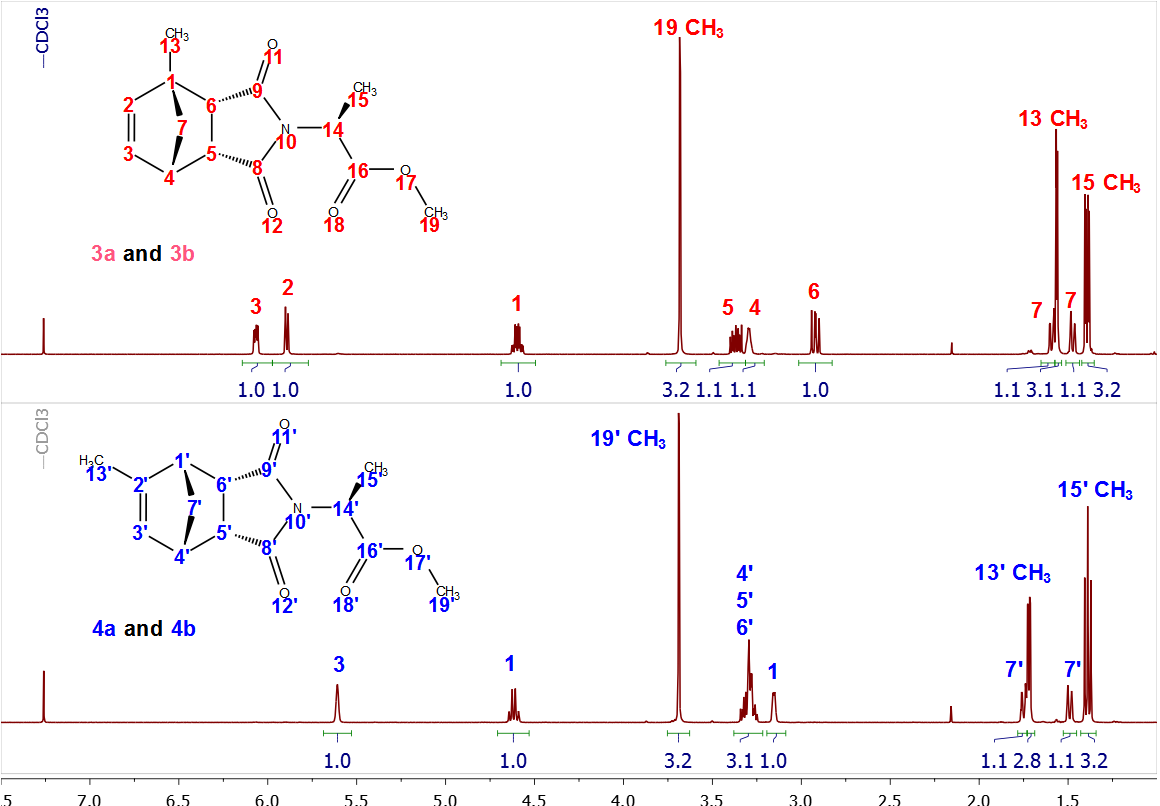 | Figure 13. Stacked plots of the 1H-NMR spectra of compound 3a, 3b and 4a, 4b |
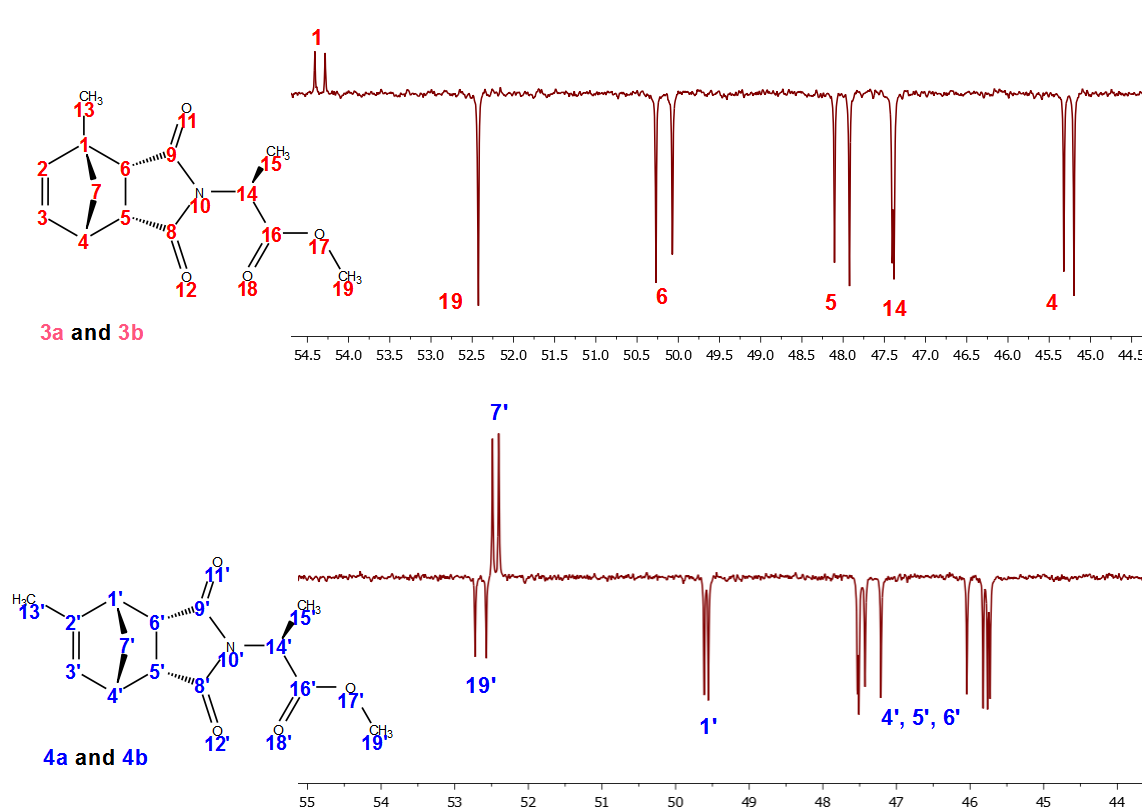 | Figure 14. Partial APT-NMR spectra of compounds 3a, 3b and 4a, 4b |
Further structural assignment could be based on various two-dimensional NMR experiments. For the diastereomeric mixture of compounds 3a and 3b, some proton signals are also duplicated and that must be accounted for in spectral interpretation. It is easy to establish the H-4 signal and from there assign H-5, H-6 and H-7 by COSY. The chemical shift assignment of H-7 can also be supported by relating H-7 to di-substituted C-7 with the help of HSQC and APT experiments. The 19-CH3 signal can be assigned by its shift to lower frequency due to the neighbouring oxygen atom and its splitting pattern i.e. singlet. The HMBC experiment shows the correlation of 19-CH3 with C-16. Since C-16 is a quaternary carbon of the carbonyl group; APT and 13C experiments could be used as well for additional confirmation. Similarly, 15-CH3 protons and C-16 have a cross peak in HMBC. The protons of 15-CH3 and H-14 are neighbours; therefore, a COSY experiment should show correlation between these two groups. The methyl groups 13-CH3 have characteristic shifts of 17.81 and 17.84 ppm in the 13C NMR spectrum. Also, HMBC experiment supports the assignment of the methyl signal with correlation between C-13 and H-2, H-7, H-6. The remaining two quaternary carbonyl carbons C-8 and C-9 have distinct positions in 13C NMR which makes APT usable for their assignment.The proton NMR spectrum of the diastereomeric mixture 4a and 4b also shows duplicated signals. The double bond proton (H-3’) signal is easy to establish due to its characteristic shift (5.61 ppm). Further, a COSY experiment could reveal the position of H-4’, H-5’ and H-6’. The assignment of H-7’ could be confirmed by COSY and additionally by HSQC and APT experiments since those are protons attached to a di-substituted carbon. The H-1’ signal determination might be challenging since in the COSY spectrum, multiple cross-peaks between H-4’, H-5’, H-6’ and H-1’ give some ambiguity in the interpretation of the spectrum. However, HMBC provides useful information about connectivity between the above-mentioned protons. The H-1’ signal should not have a cross-peak with C-2, meanwhile H-4’, H-5’ and H-6’ should have cross-peaks with C-2.The assignment of 19’-CH3, C-16’, 15’-CH3, H-14’ and C-8’, C-9’ is done in the same manner as described earlier for the mixture 3a and 3b. The 13-CH3 peaks were determined by their characteristic shifts in the carbon NMR spectrum (16.82 and 16.73 ppm). Additionally, HMBC can be used to support the methyl group assignment.
4. Conclusions
This laboratory experiment is designed to introduce undergraduate students to the structural analysis of components of inseparable reaction mixtures through direct NMR analyses. In addition, the experiment demonstrates the separation of diastereomers. Elements of regiochemical and stereochemical relationships are also featured. Using readily available chemicals and a well-studied Diels-Alder reaction, students can, within two or three lab periods, be exposed to one-and two-dimensional NMR spectroscopic techniques as well as the use of diastereomers chromatography separation to double check their prior assignments for the analysis of complex mixtures.
ACKNOWLEDGEMENTS
The authors thank Simon Fraser University (SFU) Chemistry Department for financial support, and Prof. B. Mario Pinto (SFU Chemistry) for his critical reading of this manuscript.
Supporting Information
All raw “full package” NMRs as well as labelled relevant NMR spectra, 1H-NMR correlation diagrams and i-3D renderings of the Diels Alder adducts are available as supplementary information.
References
| [1] | Williamson, K. Macroscale and Microscale Organic Experiments; Houghton-Mifflin Company: New York, 2003; pp 661-678. |
| [2] | Mayo, D. W.; Pike, R. M.; Forbes, D. C. Microscale Organic Laboratory with Multistep and Multiscale Syntheses, 5th ed.; John Wiley & Sons: New York, 2011; pp 257-269. |
| [3] | Schoffstall, A. M.; Gaddis, B.; Druelinger, M. Microscale and Miniscale Organic Chemistry Laboratory Experiments, 2nd ed.; McGraw-Hill: New York, 2004; pp 265– 269. |
| [4] | Pavia, D. L.; Lampman, G. M.; Kriz, G. S.; Engel R. G. Introduction to Organic Laboratory Techniques: A Microscale Approach, 3rd ed.; Saunders: Fort Worth, 1999; pp 405-409. |
| [5] | Csicsery, S. M. Methylcyclopentadiene Isomers. J. Org. Chem., 1960, 25, 518-521. |
| [6] | Korenevsky, V. A.; Sergeyev, N. M. Proton coupling constants in methylcyclopentadienes. J. Am. Chem. Soc., 1972, (94) 8586-8587. |
| [7] | Mstislavsky, V.I.; Korenevsky, V. A.; Sergeyev, N. M.; Solkan, V. N. Proton spin coupling constants in methylcyclopentadienes. Org. Magn. Reson., 1976, (8) 368-374. |
| [8] | Gerak, C. A. N.; Sutherland, M. L.; Field, M. J.; Woo, E. H. S.; Merbouh, N. “The use of a small chalcone spectroscopy database for the introduction of advanced spectroscopy techniques at the undergraduate level” The Chem. Educator 2016, (21), 119-128. |
| [9] | Woo, E. H. S.; Field, M. J.; Sutherland, M. L.; Gerak. C. A. N.; Merbouh, N. “A simple case study for the introduction of the HMBC and the EI-MS techniques to second and third year undergraduate students” The Chem. Ed. J. (CEJ), Vol. 18 (Serial No. 33). The date of issue: March 1, 2017. |
| [10] | Miller, V. R. Use of HSQC, HMBC, and COSY in Sophomore Organic Chemistry Lab. Nmr Spectroscopy in the Undergraduate Curriculum, 2013, 1128, 103. |
| [11] | Ford, W. T. Cycloaddition of Benzyne to Substituted Cyclopentadienes and Cyclopentadienyl Grignard Reagents. J. Org. Chem., 1971, (36) 3979-3987. |
| [12] | McLean, S.; Haynes, P. Substitution in the cyclopentadienide anion series: Polymethylation. Tetrahedron, 1965, (21) 2343-2351. |
| [13] | Breitmaier, E., Structure elucidation by NMR in organic chemistry: a practical guide. 3rd rev. ed.; Wiley: Chichester, West Sussex, England, 2002; pp 21-25. |
| [14] | Cooley, J. H.; Williams, R. V. endo- and exo-Stereochemistry in the Diels-Alder Reaction: Kinetic versus Thermodynamic Control. J. Chem. Ed., 1997, (74) 582-585. |
| [15] | Kamezawa, N.; Ueda, T. Nuclear magnetic resonance studies of methyl-5,6-dicarboxy-2-norbornene derivatives. Org. Magn. Reson., 1971, (3) 557-565. |
| [16] | Kamezawa, N.; Sakashita, K.; Hayamizu, K. Nuclear magnetic resonance studies of 5,6-dicarboxy-2-norbornene derivatives. Org. Magn. Reson., 1969, (1) 405-414. |
| [17] | Biagini, C.G.; Bush, S. M.; Gibson, S. C.; Mazzariol, V.; North, L.; Teasdale, M. G.; Williams, C. M.; Zagotto, G.; Zamuner, D. The Synthesis of N-Norbornenyl-Amino Acids and Esters: Monomers for the Preparation of Well Defined Polymers. Tetrahedron 1995. 51. 7247-7262. |
| [18] | Gunkara, O.; Kulu, I.; Nuket, O.; Kaufmann, D. E. Synthesis of Arylated Norbornyl Amino Acid Esters. Monatsh Chem. 2010, 141, 1237-1243. |

 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML