-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Journal of Laboratory Chemical Education
p-ISSN: 2331-7450 e-ISSN: 2331-7469
2017; 5(4): 55-66
doi:10.5923/j.jlce.20170504.01

Study of UV Filters as an in silico (QSAR) Graduate Project
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLD. González-Arjona1, G. López-Pérez1, A. Gustavo González2, M. M. Domínguez1, W. H. Mulder3
1Department of Physical Chemistry, Universidad de Sevilla, Sevilla, Spain
2Department of Analytical Chemistry, Universidad de Sevilla, Sevilla, Spain
3Department of Chemistry, University of the West Indies, Kingston, Jamaica
Correspondence to: D. González-Arjona, Department of Physical Chemistry, Universidad de Sevilla, Sevilla, Spain.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

A simplified in silico QSAR study, involving more than 50 molecules that are commonly used as UV filters in sunscreens, is presented. Details of methodology are described and illustrated step by step and the use of available tools is demonstrated, including data set selection, generation of molecular structure files, selection of computational methods, assessment of the relative importance of molecular properties, generation of theoretical UV-Vis spectra and construction of a QSAR model. With the aim of introducing the QSAR methodology at undergraduate level, some simplifications have been made. Multiple Linear Regression (MLR) has been selected as a modeling strategy to correlate the molecular descriptors and the sunlight blocking effect. The full area under the UV-Vis spectrum has been used as a simple measure of the protection factor. Theoretical UV-Vis spectra have been generated by using the configuration interaction (CI) approximation. The ground state molecular geometry has been optimized by the semi-empirical ZINDO computational method. Inclusion of a solvent model in the optimization of molecular geometry to account for solvatochromic effects has not been considered. On the other hand, total number of rings, number of aromatic rings, molecular dipole moment and HOMO/LUMO energy gap, are considered to be significant parameters in the MRL model. The HOMO/LUMO energy gap is the variable of choice when optimized single linear regression analysis is carried out separately for each of the functional groups of the compounds. The great variety of tools and procedures used in building QSAR models has been found to provide ample scope for active involvement by the students, exposing them to a fairly broad array of modern methods in areas like drug discovery and toxicology.
Keywords: QSAR, Sunscreens, UV-VIS absorption, Graduation projects
Cite this paper: D. González-Arjona, G. López-Pérez, A. Gustavo González, M. M. Domínguez, W. H. Mulder, Study of UV Filters as an in silico (QSAR) Graduate Project, Journal of Laboratory Chemical Education, Vol. 5 No. 4, 2017, pp. 55-66. doi: 10.5923/j.jlce.20170504.01.
Article Outline
1. Introduction
- More than half a century has passed since Hansch et al [1] laid the foundations of the modern methodology of “Quantitative Structure-Activity Relationships” (QSAR), initially building on the work of Polanyi and subsequently Hammett who introduced the concept of “Linear Free Energy Relationships” (LFERs), by correlating the chemical structure of the phenoxyacetic acids with their biological activity in plant growth. Since then, QSAR has evolved to a point where it provides reliable, statistically predictive models.Designing a chemical compound that possesses certain properties is usually very complicated relative to the effort involved in synthesizing a particular molecular structure. Although it may be relatively easy to deduce a reaction mechanism from a knowledge of molecular structure, it is often far more difficult to predict its properties based on this structure. Whereas the reactivity appears as an intrinsic characteristic of the molecule this is not necessarily true for physicochemical or biological properties. The peculiar properties and behavior of a compound depend on both its internal structure and the molecular environment in which it finds itself. The surroundings interact with the entire molecule and more strongly so with specific sites. However, unlike chemical reactions, these interactions are relatively weak and do not lead to the breaking or formation of chemical bonds. Therefore, the modeling needed to relate the structure and a particular activity quantitatively, QSAR, is not a trivial task. The use of QSAR models for the screening of chemical/biological products is a matter of great interest in both the chemical and pharmaceutical industry, toxicology, medicine, environmental studies, etc. The QSAR modeling method appears as one of the most useful tools in the understanding and prediction of properties and effects of chemical/biological compounds in the areas of environment and health. This methodology constitutes an efficient and operative means of assessing chemical hazards. In addition, the use of QSAR offers other advantages, such as low cost, high degree of investment recovery and it may even reduce the need for using animals in laboratory testing of new drugs. All these applications have attracted the growing interest from government agencies for in silico predictions [2, 3]. Clearly, QSAR modeling is a multidisciplinary subject, which combines aspects from computer science, chemistry, statistics (cheminformatics) and knowledge of disciplines, particularly toxicology, that explore the activity of chemicals as it relates to health and environment. The mechanism underlying the protection by sunscreens is well known. It primarily involves absorption of UV light by electrons in a molecule’s frontier orbital [4]. Thus, the active compound in sunscreens protects the skin from sunburn by blocking UV radiation. The extent of this action can be evaluated experimentally based on the UV-Vis absorption spectrum. Therefore, a knowledge of the nature of UV filter action, the straightforward identification of a target function (UV-Vis spectral area), together with the relative ease with which molecules of intermediate size can be modeled, makes the group of molecules that act as UV filters an ideal object for a QSAR-type study.With the aim of introducing the QSAR methodology at undergraduate level, this paper describes a simplified in silico QSAR study for different UV filters. The model is developed using calculated numerical molecular descriptors that encode information about each molecular structure. From the different modeling strategies, the simple Multiple Linear Regression (MLR) has been selected to correlate the molecular descriptors and the sunlight blocking effect, in this case the full area under the UV-Vis spectrum. In order to allow completion of the project within a reasonable time, some simplifications have been made to obtain the molecular descriptors. Thus, the theoretical method employed to generate both the molecular descriptors and the UV-Vis spectral area are based on semi-empirical quantum chemical methods. Moreover, the solvent effect is not taken into account [5]. Modeling of the solvatochromic effect is a complex undertaking and is outside the didactical scope of the present paper [6]. Therefore, the conclusions that can be drawn are somewhat limited. Notwithstanding, the basic steps in QSAR methodology, viz., the building up and optimization of 3D molecular structures, generation and selection of descriptors and model construction and validation are demonstrated and analyzed.This paper therefore explores the quantitative relationship between absorption in the UV-Vis region with the chemical structure of UV filters, without taking into account possible spectral shifts due to the presence of a solvent.
2. Methodology
- The sun protection mechanism using organic chemicals is based on the strong absorption of UV light by these molecules. Therefore, the molecular energy gap for the frontier orbital set close to HOMO-LUMO should have a value in the UV-Vis range. This energy gap can be realized with aromatic compounds having conjugated C=C double bonds and other diverse functional groups. The UV-Vis absorption interval and strength can be modified by varying the type and position of molecular substituents. The entire project can be developed in several stages.
2.1. Data Set Selection
- The selection of compounds that constitute the data set should be the first step of the project. Mainly seven different classes of organic compounds are commonly employed as active UV filters, Figure 1, shows their basic schematic structures. The compound data set for this study was selected from the globally approved list of UV-Vis filters [7, 8].
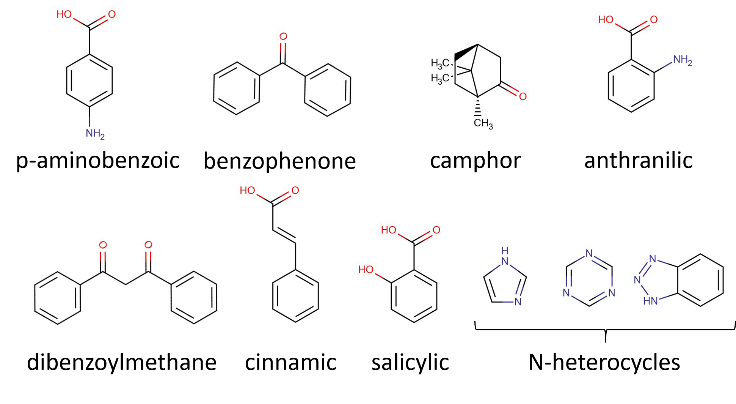 | Figure 1. Schematics structures of different classes of organic compounds, containing conjugated double bonds, frequently used as UV filters |
2.2. Molecular Computer File Generation
- For each compound, a computer file containing the molecular structure readable by computational molecular programs should be generated. Common molecular files read by the majority of the computational chemistry programs are the mol file, MDL and structure data file, SDF [9]. Computational Chemistry for Chemistry Educators [10], offers information and resources for molecular modeling, with systematic instructions for molecular modeling with different softwares. From the name of each organic compound, a computer file was generated containing the 3D initial molecular structure. Different web resources can be employed to obtain the text file containing the molecular structure, from the IUPAC name and/or using the “simplified molecular-input line-entry system”, SMILES [11]. To avoid issues arising from different nomenclature and/or isotopes, each compound is identified by its unique CAS registration number [12]. This registry can be obtained from CACTUS (CADD Group Chemoinformatics Tools and User Services) [13]. From CAS a standard molecular file can be exported using the ChemSpider [14], ChemCell [15], now also available on Google Spreadsheets, or Chemicalize [16] web resources. Among them, ChemSpider, Chemicalize and PubChem are recommended, they offer web links that give access to estimates of assorted molecular properties, or descriptors in QSAR nomenclature, from its structure: ACD/Labs [17], EPISuite [18], ChemAxon [19] and Mcule [20].
2.3. Theoretical UV-Vis Spectra and Selection of Computational Method
- The sunblock protection efficiency is usually characterized by a test in vivo. Estimation of the sun protection factor, SPF, is based on the ratio of the minimum erythematic dose between protected and unprotected skin. The comparable parameter used in vitro does not have a unique and widely accepted definition. In this paper, the modified parameter defined by UV area per unit of wavelength [21], recommended based on FDA regulations [7], is used:
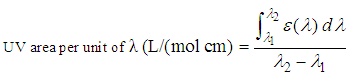 | (1) |
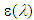 is the extinction coefficient and
is the extinction coefficient and 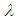 is the wavelength. This target parameter can be easily estimated from experimental or theoretically generated UV-Vis spectra.The theoretical UV-spectrum can be generated by computational methods using the approximation known as configuration interaction, CI, [22]. The transition energy between the ground and the excited states are computed for the same geometry, that of the ground state. This energy is given usually as a wavelength for each allowed electronic transition. The relative intensity for each transition is related to the change in the dipolar moment strength and is reported as the oscillator strength for each electronic transition [23]. Forbidden transitions have an oscillator strength value close to zero. For each active transition (i), the extinction coefficient is produced as a Gaussian band shape, with a constant half-width (approx. 0.4 eV) by using the method proposed in Gaussian Tech Notes [24]:
is the wavelength. This target parameter can be easily estimated from experimental or theoretically generated UV-Vis spectra.The theoretical UV-spectrum can be generated by computational methods using the approximation known as configuration interaction, CI, [22]. The transition energy between the ground and the excited states are computed for the same geometry, that of the ground state. This energy is given usually as a wavelength for each allowed electronic transition. The relative intensity for each transition is related to the change in the dipolar moment strength and is reported as the oscillator strength for each electronic transition [23]. Forbidden transitions have an oscillator strength value close to zero. For each active transition (i), the extinction coefficient is produced as a Gaussian band shape, with a constant half-width (approx. 0.4 eV) by using the method proposed in Gaussian Tech Notes [24]: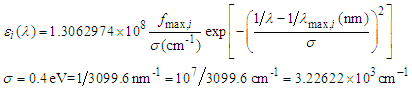 | (2) |
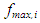 and
and 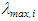 are the oscillator strength and the wavelength (nm) of the electronic state i, respectively.The full spectrum can be easily convoluted by adding all active transitions in the UV-Vis energy interval:
are the oscillator strength and the wavelength (nm) of the electronic state i, respectively.The full spectrum can be easily convoluted by adding all active transitions in the UV-Vis energy interval: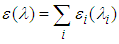 | (3) |
2.4. Estimating Molecular Properties
- The ability to predict properties depends strongly on an appropriate choice of descriptors. There is a variety of molecular properties that can be used in QSAR. They can be classified as physicochemical [27, 28] and topological [29], so that in turn they can be divided into groups [30-32]:• Constitutional (number and type of atoms, rings, MW, ...)• Topological (indices that represent the structure using graphs)• Geometric (Areas, volumes, ...)• Mechanical (moments of inertia, …)• Electrostatic (polarizability, partial charge, dipole moment, ...)• Quantum (frontier molecular orbitals, free valence, bond order, electronic energy, electrostatic interaction, ...)• Thermodynamic (heat of formation, heat capacity, ...)Nevertheless, there is a wide variety of software applications to obtain molecular descriptors: Codessa Pro [33], Dragon 6 [34], CORINA Symphony [35], and Molecular Operating Environment [36]. Most of them provide more than 1000 descriptors for any molecule. However, the use of a large number of descriptors in the construction of the model has, in addition to increasing computing time, two disadvantages: increase of ‘noise’, due to the use of some correlated descriptors, and model ‘over-training’. The latter tend to produce models with low predictive power. Nevertheless, most of the above-mentioned software provides tools for fast data screening to obtain the most significant descriptors, minimizing the above-mentioned adverse effects.
2.5. QSAR Model Construction
- The first stage in the model building process will be the selection of variables (properties) that could contribute significantly to the model as well as the estimation of their level of importance. These properties can be grouped in matrix form, where each column is associated with a specific property and each row corresponds to a chemical compound. These properties are considered as independent variables, termed an X matrix. An additional column is incorporated containing the dependent variable: a molecular property (experimentally determined or theoretically calculated) to be replicated by the model, the Y matrix. The analysis of this matrix equation is performed depending on the types of variables and their mathematical relationship. Thus, the following types of fundamental modeling strategies have been considered in the chemometric literature [37, 38]:• MLR, Multiple Linear Regression• PCR, Principal Component Regression• PLS, Partial Least Squares• TFA, Target Factor Analysis• ANN, Artificial Neural Networks• SVM, Support Vector MachinesIf it can be assumed that the selected independent properties mainly contribute linearly to the model, the multiple linear regression (MLR) based on the least squares method should be the model technique of choice. This model can only be applied when the variables have low synergy among them, and no quadratic terms need to be considered. With a view to the pedagogical scope of the project, students mainly employed MLR which can also be easily handled by Excel. Therefore, the MLR model equation, in matrix format, will be:
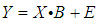 | (4) |
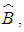 the response matrix
the response matrix 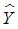 and residual error matrix can be estimated by least squares analysis:
and residual error matrix can be estimated by least squares analysis: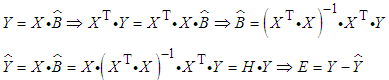 | (5) |
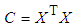 | (6) |
3. Results and Discussion
- After compound selection, the molecular file with the molecular structure was generated from the web facilities described above. In the present project the CAChe 7.5 software as “all-in-one” [42] has been employed. CAChe, computer-aided chemistry, is a 3D molecular modeling tool environment capable to build up molecules. The program performs molecular calculations, named “experiments”, using classical and quantum mechanical theories providing electronic properties of the optimized molecular geometry. Moreover, the software provides a separate “ProjectLeader”, as spreadsheet interface to perform batch-processing calculations for several chemical samples at the same time (structures, properties, statistical analyses, multiple linear regression, calculations based on customizable equations …). This separate tool is specially adequate and comfortable to perform QSAR studies. Using this spreadsheet interface the table with the molecular properties for each molecule can be built up.
|

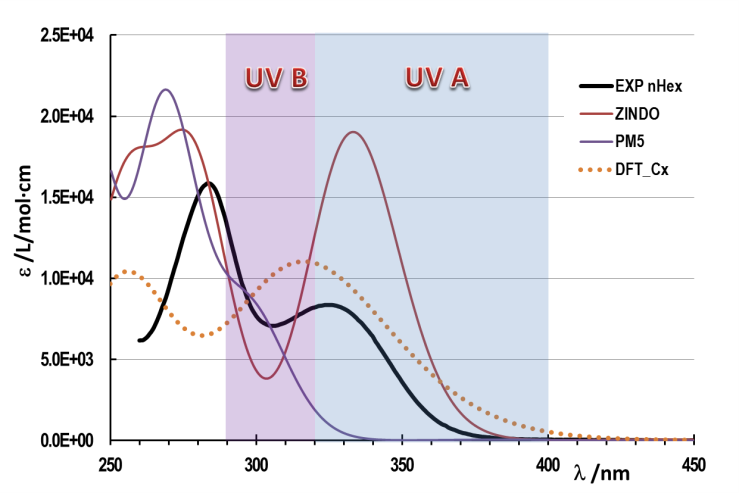 | Figure 2. Oxybenzone experimental UV-Vis spectrum in n-Hexane compared with that theoretically calculated using different computational methods ZINDO and PM5 in vacuum and DFT in cyclohexane, modelled as a continuum with uniform dielectric constant |
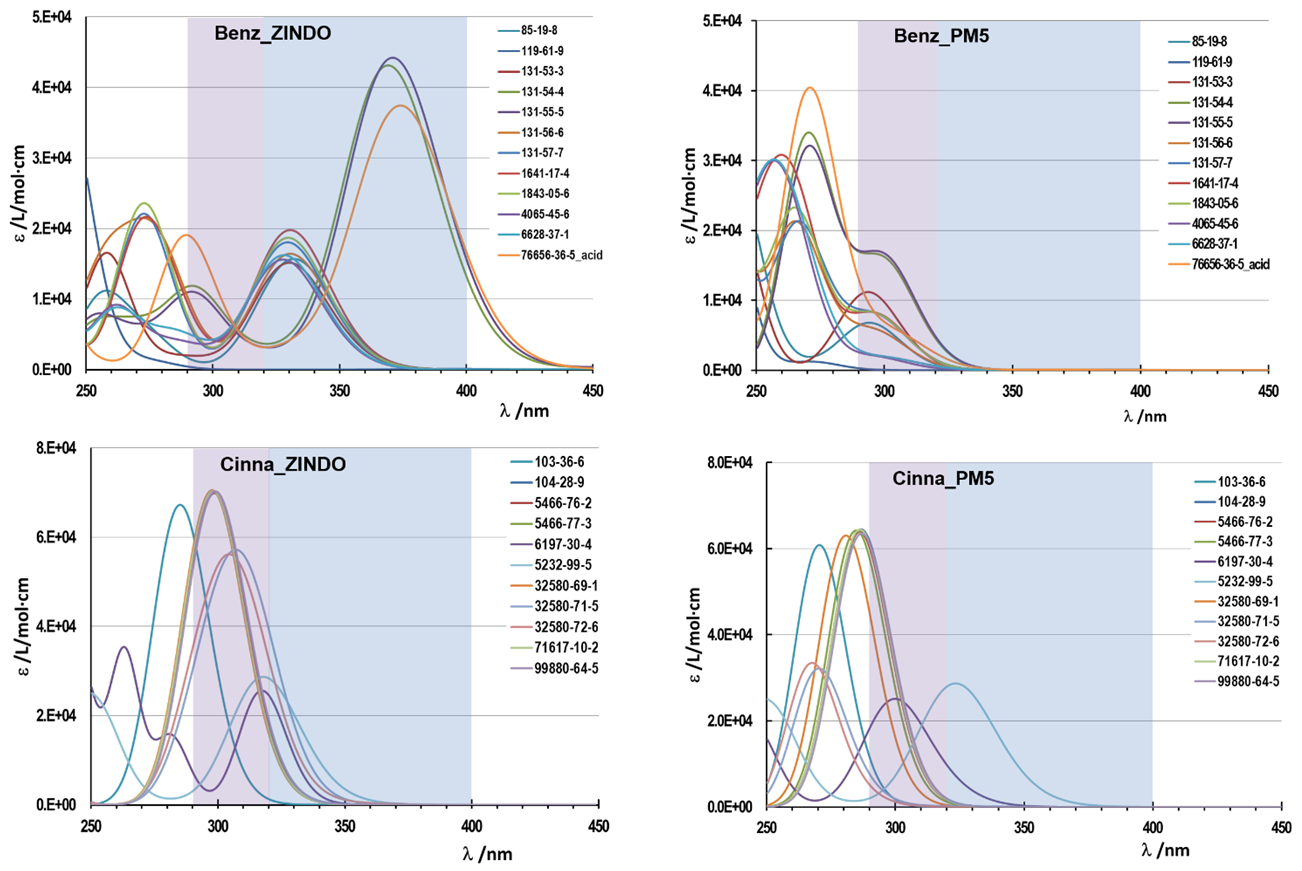 | Figure 3. UV-Vis spectra of Benzophenones (Benz) and Cinnamates (Cinna) obtained with different semi-empirical computational methods: left ZINDO, right PM5. Each compound is identified by the CAS # |
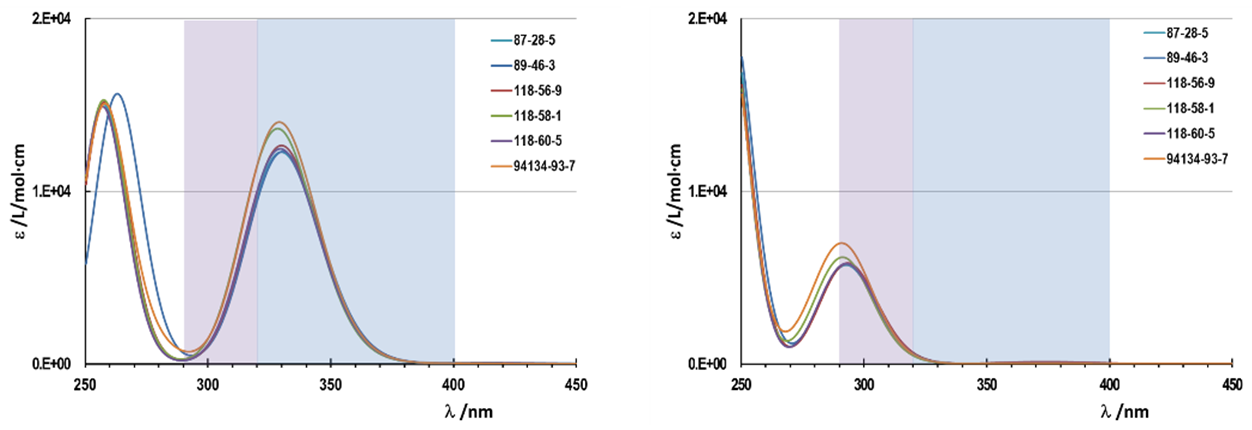 | Figure 4. UV-Vis spectra for salicylates obtained by ZINDO with different geometry. Left, spectra with intramolecular H-bond between the oxygen of the carbonyl group and the hydrogen of the phenol group. Right, spectra without intramolecular H-bond |
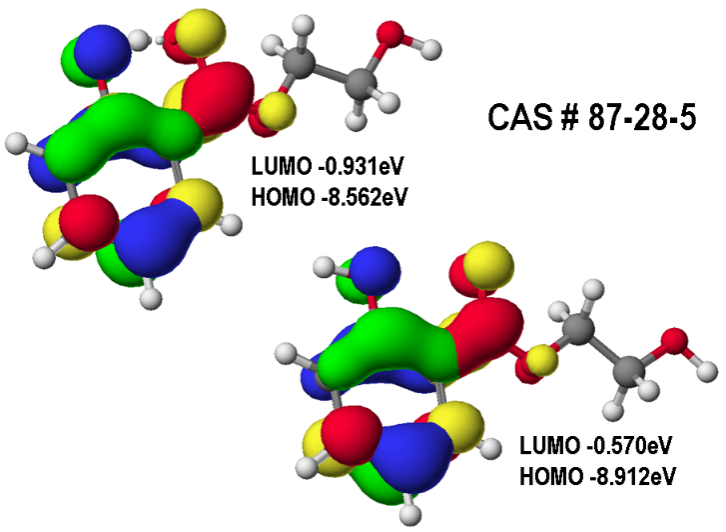 | Figure 5. Frontier orbitals shown as electron isodensity surfaces (HOMO (blue-green), LUMO (red-yellow)) for 2-hydroxyethyl salicylate with and without intramolecular H bond, _C=O···H—O_ |
3.1. MLR Model Building
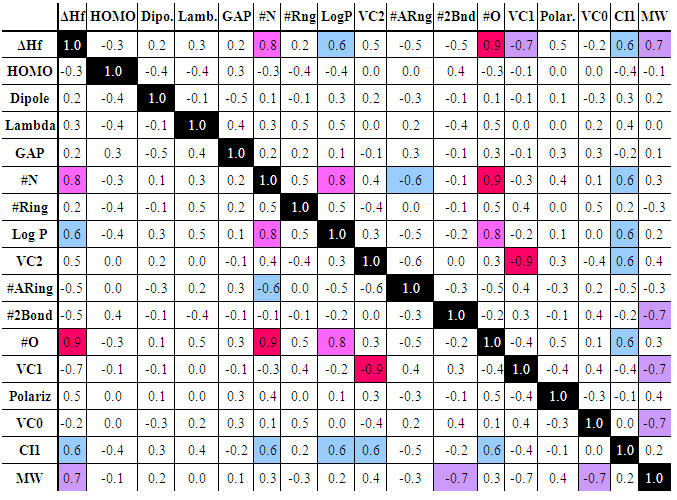
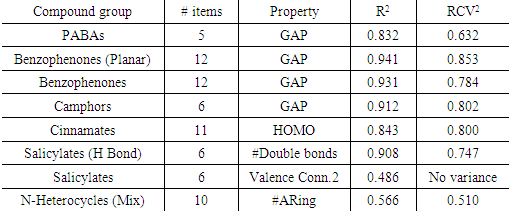
- Before starting the modeling, close data scrutiny will have to be the first step. Simple statistical analysis (number of valid data, mean, standard deviation…) can help to detect possible codification errors and outliers. The use of the “box and whisker” plots is recommended for discovering spurious and suspicious values, ultimately simplifying and improving the analysis.Thus, in the actual statistical analysis of a data set, when considering the property molecular weight (MW), compounds with values exceeding 500 Daltons should be considered as suspicious outliers. Additionally, those molecular properties that have a close relationship with the MW will also have suspicious values for this group of compounds. However, this group should not be removed from the data set because they represent the new generation of sunscreen compounds. Compounds obeying the 500 Da rule have lower skin absorption and toxicity level [47], and so, they are preferred in the sunscreen formulation.Another important preliminary and necessary analysis is the inspection of the correlation matrix, Eqn. 6. This matrix can be easily obtained from the spreadsheet matrix multiply function between the transposed matrix of molecular properties and the original molecular properties matrix.Table 2, gathers the correlation values for some molecular properties. To facilitate their analysis, cells with a correlation level greater than 0.6 have been given different colors. Thus, some molecular properties show a moderate degree of correlation with several other: MW, Log P, #Nitrogen, #Oxygen, Connectivity Index 1, Heat of formation...
|
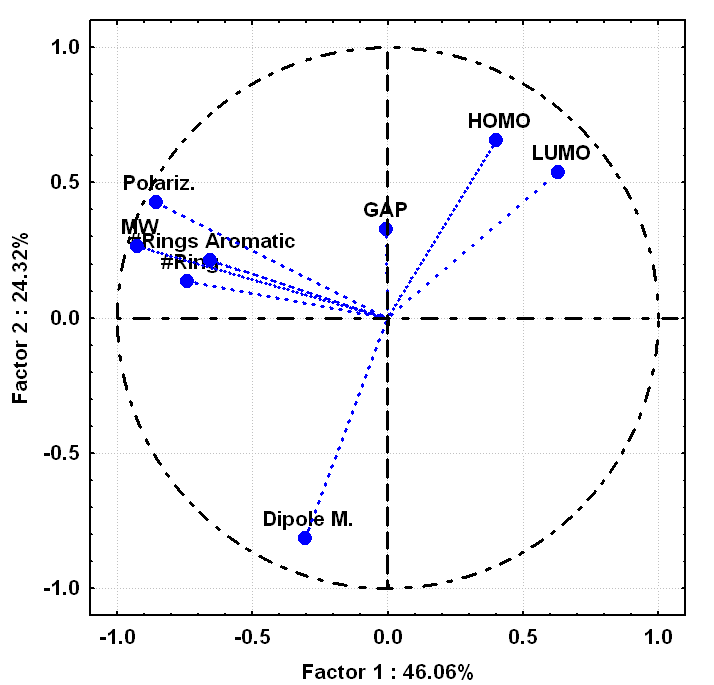 | Figure 6. Projections of the molecular properties (variables) on the two first principal components plane |
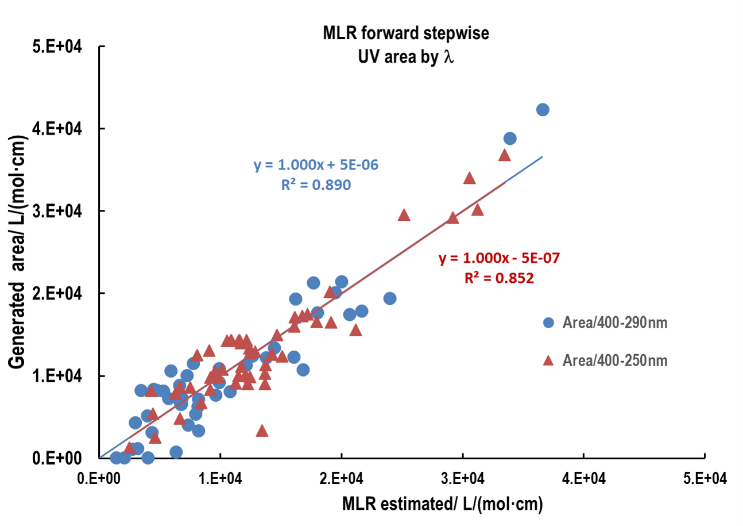 | Figure 7. Sunscreen scatter plot of UV area by wavelength of theoretically generated (ZINDO) versus the corresponding areas estimated by MLR forward stepwise. Blue dots correspond for the area calculated for the spectral range from 290nm to 400nm and red triangles the area calculated for the 250nm to 400nm interval |
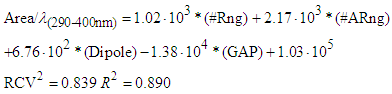 | (7) |
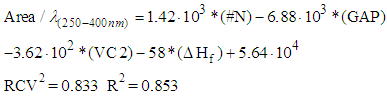 | (8) |
|
4. Conclusions
- Modeling in silico UV-Vis spectra can be performed by the configuration interaction singles (CI-s), a relatively low-level computational method for computing excited states. The project ambit, graduate level, simple organic molecules, together with the possible computational limitations, motivated the selection of a semi-empirical theoretical method for molecular modeling. Moreover, the UV-Vis spectrum is not influenced only by the compound itself but also by its environment, such as the solvent, solution pH, etc. These facts introduce some degree of difficulty that have prevented QSAR studies from being more widely used in chemical education. Nevertheless, among the various semi-empirical methods, ZINDO has parameters designed to match the UV-Vis spectra. Despite the shift of the maxima in the case of some spectra, the correct overall trend for the full UV-Vis spectra of simple organic molecules can be obtained. Additionally, the 2D-QSAR study is mainly concerned with the correlation between the simple molecular structure descriptors and the UV-Vis spectral area. Thus, in spite of using approximate molecular modeling, the application of this methodology has a reasonable basis. QSAR analysis compares relative contributions and it is expected that the method employed provides approximately the same shifts for all molecules studied.Therefore, this in silico QSAR study of sunscreens can be a good starting point to introduce the QSAR methodology at the final year undergraduate level. Nevertheless, limitations of its applicability should be clearly pointed out.
ACKNOWLEDGEMENTS
- The authors are very grateful to Prof. R. Rodríguez Pappalardo for his helpful comments and suggestions. We also thank to students of Chemistry: A. Martínez Pascual and A. Palacios Morillo for their dedication in developing part of the “Sunscreen Project”.
Appendix 1
- In silico QSAR UV filters study
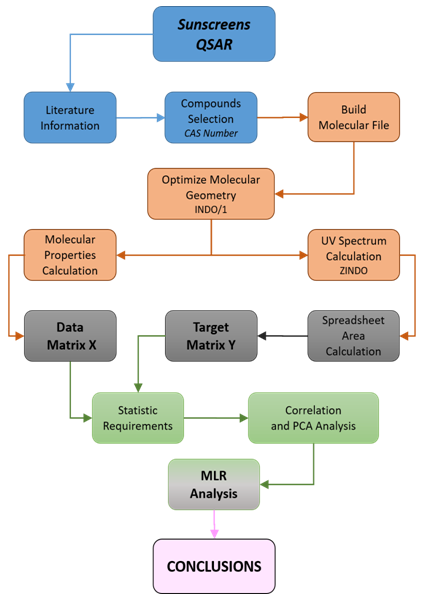 Ÿ QSAR topic selection (Blue section).ο Update literature, SciFinder, Google scholar,…ο Compounds collection, ref. 21 and literature cited therein, FDA regulations,…§ Unequivocally identification by CAS Registry Number, ref 12.Ÿ Molecular modeling tool. CAChe, ref 42 (Orange section).ο Molecular structure file generation.§ From CAS registry #, the references 14 and 16 provide an initial molecular file to be read by CAChe.§ Ground state molecular geometry is optimized by semi-empirical method INDO/1.§ Estimation of molecular properties (Data matrix X), using CAChe ProjectLeader. Export to Excel format.ο UV spectra generation.§ Generate UV spectrum using Configuration Interaction, ref 22, and ZINDO, ref. 44.§ Export a text file containing the UV-Vis spectrum at 1nm interval for each compound.Ÿ Spreadsheet calculations (Grey section).ο Estimation of the UV area per unit of wavelength, eq. 1. The integral is estimated by using the trapezoidal rule (Target matrix Y).Ÿ Statistical calculations (Excel or statistical package, references 40 and 41) (Green section). ο Initial matrix data scrutiny: codification errors, outliers values,…ο Correlation matrix inspection, drop highly correlated molecular properties. ο Principal Component Analysis, projection of molecular properties on factors space and estimation of maximum number of variables.ο Modeling strategy selection: Multi Linear Regression (MLR).§ Checking variable (molecular properties) for normal distribution and homoscedasticity.§ Molecular properties selection in the model by using MRL forward stepwise.§ Model cross-validation (not performed in this study).§ Best simple linear regression for each class of organic UV filter.Ÿ Conclusions (Pink section)ο Advantages and disadvantages of QSAR.
Ÿ QSAR topic selection (Blue section).ο Update literature, SciFinder, Google scholar,…ο Compounds collection, ref. 21 and literature cited therein, FDA regulations,…§ Unequivocally identification by CAS Registry Number, ref 12.Ÿ Molecular modeling tool. CAChe, ref 42 (Orange section).ο Molecular structure file generation.§ From CAS registry #, the references 14 and 16 provide an initial molecular file to be read by CAChe.§ Ground state molecular geometry is optimized by semi-empirical method INDO/1.§ Estimation of molecular properties (Data matrix X), using CAChe ProjectLeader. Export to Excel format.ο UV spectra generation.§ Generate UV spectrum using Configuration Interaction, ref 22, and ZINDO, ref. 44.§ Export a text file containing the UV-Vis spectrum at 1nm interval for each compound.Ÿ Spreadsheet calculations (Grey section).ο Estimation of the UV area per unit of wavelength, eq. 1. The integral is estimated by using the trapezoidal rule (Target matrix Y).Ÿ Statistical calculations (Excel or statistical package, references 40 and 41) (Green section). ο Initial matrix data scrutiny: codification errors, outliers values,…ο Correlation matrix inspection, drop highly correlated molecular properties. ο Principal Component Analysis, projection of molecular properties on factors space and estimation of maximum number of variables.ο Modeling strategy selection: Multi Linear Regression (MLR).§ Checking variable (molecular properties) for normal distribution and homoscedasticity.§ Molecular properties selection in the model by using MRL forward stepwise.§ Model cross-validation (not performed in this study).§ Best simple linear regression for each class of organic UV filter.Ÿ Conclusions (Pink section)ο Advantages and disadvantages of QSAR.