-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Journal of Laboratory Chemical Education
2016; 4(1): 4-8
doi:10.5923/j.jlce.20160401.02

Spectroscopic Characterization of the Tertiary Amine, N-(5-Hydroxypentyl)-N-(3aminopropyl)dodecanamide (1)
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAbdalla M. Walwil
Department of Chemistry, Arab American University, Jenin, Palestine
Correspondence to: Abdalla M. Walwil, Department of Chemistry, Arab American University, Jenin, Palestine.
| Email: |  |
Copyright © 2016 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

We are presenting educational work that could be implemented in chemistry teaching labs on the process of pyramidal inversion, also known as nitrogen inversion. Compounds 1and 2, N-(5-Hydroxypentyl)-N-(3-aminopropyl)hexanamide,andN-(5-Hydroxypentyl)-N-(3-aminopropyl)dodecanamide respectively were prepared as two intermediates during the synthesis of the natural product Acarnidine 3, N-[5-(guanidinopentyl)-N-[3-[(3-methyl-1-oxo-2-butenyl)amino]propyldodecanamide and its analog 3a,N-[5-(guanidinopentyl)-N-[3-[(3-methyl-1-oxo-2-butenyl)amino]propylhexanamide.The starting materials 4, 5-aminopentanol,and 4a, acrylonitrile, were converted to 3 and 3a in six steps. Compounds 1 and 2, found as mixtures of rotamers (1a & 1b) and (2a & 2b) respectively, were produced from their precursors 5 and 6 respectively. The 1H-NMR spectra for compound 1, in DMSO-d6 at 20 and 75°C or in CDCl3, are presented here and analyzed. Here I am trying to present the basic phenomenon, sometimes overlooked by many researchers, which occurred to certain nitrogen-containing compounds known as pyramidal or nitrogen inversion. 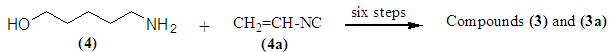
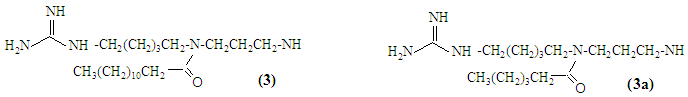
Keywords: Pyramidal inversion, Nitrogen inversion, N-(5-Hydroxypentyl)-N-(3-aminopropyl)hexanamide, and N-(5-Hydroxypentyl)-N-(3-aminopropyl)dodecanamide
Cite this paper: Abdalla M. Walwil, Spectroscopic Characterization of the Tertiary Amine, N-(5-Hydroxypentyl)-N-(3aminopropyl)dodecanamide (1), Journal of Laboratory Chemical Education, Vol. 4 No. 1, 2016, pp. 4-8. doi: 10.5923/j.jlce.20160401.02.
Article Outline
1. Introduction
- The nitrile functional groups of compounds 5 and 6 were selectively reduced to the corresponding primary amines 1 and 2, respectively, in the presence of a mixture of sodium borohydride and cobalt (II) hexahydrate in 10% ethanolic ammonia [1-5]. Ammonia is used to drive the equilibrium backward and to prevent forming a secondary amine as a side product. In general, the side product may be avoided by adding either acetic anhydride (removes the primary amine as soon as it is formed) or excess of ammonia. Compounds 1 and 2 found to be white solids, m.p. 42-44°C and 40-42°C respectively [1]. At first glance, the target molecules 1 and 2 presented a puzzle because the two conformational isomers for 1, (1a & 1b) and the two conformational isomers for 2, (2a & 2b) did not equilibrate rapidly; therefore giving the appearance in the CDCl3 1H-NMR spectrum of substantial impurities. However, in DMSO-d6 as well as in pyridine-d5 at room temperature or at high temperature, the spectrum reduced to the simple expected form, verifying the structure [6]. Apparently, in both deuterated solvents at 75°C, DMSO-d6 and pyridine-d5, the equilibrium is fast enough and simple expected spectrum was obtained, while in CDCl3, DMSO-d6, or in pyridine-d5 at 20 C0, the equilibrium is less rapid allowing the 300 MHz NMR instrument to read both enantiomeric structures, hence, a more complex 1H-NMR is obtained. A striking example of such conformational isomerism in an amide is heterocycle 7, found as two crystalline amide rotamers (enantiomers) which are isolable at room temperature [7]. The 1H-NMR spectrum of a mixture of 7 showed doubling of all the expected signals, and at 120°C in DMSO-d6 these signals did not coalesce, which means a strongly hindered amide bond rotation of two distinct structures, 7a and 7b.
 In general tertiary-chiral amine, amine with three different alky groups, are found in enantiomeric forms. The two enantiomers are reported to be difficult to separate at room temperature because they interconvert rapidly. This interconversion process is known as pyramidal or nitrogen inversion. The barrier to the interconversion for most simple amines [8] is reported to be about 6 kcal mol-1 while the barrier energy of compound 7 inversions is reported to be 25 Kcal/mol. The salt of amine, quaternary ammonium salts, however, can be separated since they cannot undergo inversion due to the absence of the unshared pair of electrons.
In general tertiary-chiral amine, amine with three different alky groups, are found in enantiomeric forms. The two enantiomers are reported to be difficult to separate at room temperature because they interconvert rapidly. This interconversion process is known as pyramidal or nitrogen inversion. The barrier to the interconversion for most simple amines [8] is reported to be about 6 kcal mol-1 while the barrier energy of compound 7 inversions is reported to be 25 Kcal/mol. The salt of amine, quaternary ammonium salts, however, can be separated since they cannot undergo inversion due to the absence of the unshared pair of electrons. 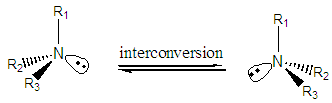 The tertiary amines (2a & 2b) enantiomeric forms expected to have higher barrier energy than that of simple ones due to the bulky R groups around the central nitrogen atom. Hence, 2a & 2b should be separable at lower temperature.
The tertiary amines (2a & 2b) enantiomeric forms expected to have higher barrier energy than that of simple ones due to the bulky R groups around the central nitrogen atom. Hence, 2a & 2b should be separable at lower temperature. 2. Experimental Section
- 1H-NMR spectra were determined with a Bruker AC-E 300 spectrometer using CDCl3 and DMSO-d6 solvents. Flash column chromatography utilized Merck 300-400 mesh. Gravity column chromatography was performed on silica gel 100-200 mesh. TLC spots were visualized using UV light. Melting points were taken on a Mel - Temp apparatus. 3-(5-Hydroxypentylamino)propanenitrile (3 or 3a). A sample of 4.03 g Acrylonitrile was added dropwise to 3.00 g of 5-aminopentanol in 15 mL of dry THF in cold media. Thhe mixture was stirred at room temperature for 4 hours. The solvent and any excess of acrylonitrile were removed by rotary evaporation to give pasty crude residue. The residue was passed into column chromatography on alumina with methanol as the eluent to give a colorless oily pure 3 in 90% yield. TLC (methanol) Rf 0.51; IR (TLF, thin liquid film) 3300, 2250 cm-1; 1H-NMR (CDCl3) δ 3.62 (2H, t, CH2-O), 2.30 (2H, t, CH2-N), 2.70 (2H, t, CH2-N), 2.58 (2H, t, CH2-CN), 2.16-1.8 (2H, br, NH & OH), 1.57 (4H, m, CH2), 1.42 (2H, m, C-CH2-C).N-(5-Hydroxypentyl)-N-(2-cyanoethyl)dodecanamide (4). A 1.71 g sample of 3 and 0.80 g of triethylamine in 15 mL of methylene chloride was added drop-wise to a solution of 1.44 g of dodecanoyl chloride in 3 mL of methylene chloride at 0°C. The reaction mixture was stirred for 3 hours and then treated with 5% HCl. The mixture was extracted with chloroform, and the organic layer was dried over CaCl2. Upon removing the solvent by rotary evaporation, a light yellow oily residue was produced. The residue was chromatographed on silica gel with ethyl acetate as the eluent to give a colorless oily product in 85% yield. TLC (2:10 EtOH:EtOAc) Rf 0.40; IR (neat) 3430, 2248, 1635, 1380 cm-1; 1H-NMR (CDCl3) δ 3.66 (2H, t, CH2-O), 3.57 (2H, t, CH2-NCO), 3.00 (2H, t, CH2-NCO), 2.72 (2H, t, CH2-CN), 2.33 (2H, t, CH2-CO), 1.66 (6H, m, CH2), 2.41-1.32 (1H, s, OH), 1.24 (18H, m, CH2), 0.92 (3H, t, CH3).
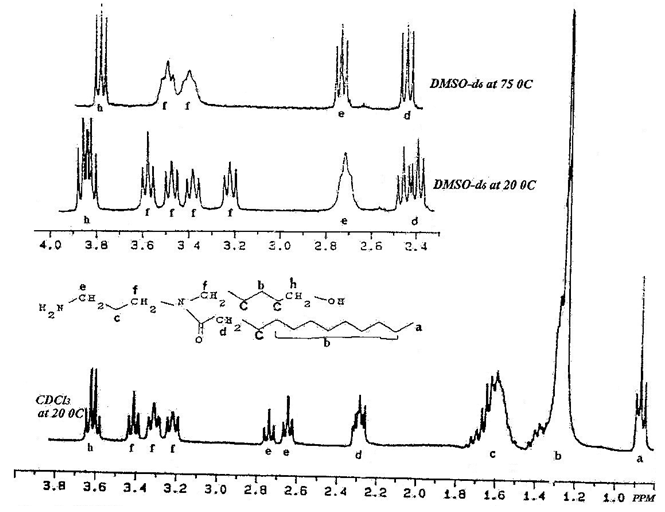 | Figure 1. 1H-NMR spectra of compound 1 in deuterated chloroform 20 °C and in deuterared dimethyl sulfoxide at 20 & 75 °C |
- N-(5-Hydroxypentyl)-N-(3-aminopropyl)dodecanamide (5). A sample of 2.9 g of sodium borohydride was added in portions to a mixture of 2.2 g of compound 4 and 3.8 g of cobalt (II) chloride hexahydrate in 40 mL of 10% ethanolic ammonia (10 mL of concentrated NH4OH and 90 mL ethanol) with stirring at room temperature for four hours. After removal of ethanol by rotary evaporation and unreacted nitrile by extraction with methylene chloride, the aqueous layer was made alkaline with concentrated ammonium hydroxide solution and then extracted with three 20-mLportions of methylene chloride. The extracts were rotovapped to yield a solid residue which was purified by flash chromatography on silica gel with 1:1 EtOH:EtOAc to give a white solid, mp 42-44°C, in 75% yield. TLC (2:10 EtOH:EtOAc) Rf 0.59; IR (KBr) 3350, 1638, 1380 cm-1; 1H-NMR (DMSO-d6, at 20°C) δ 3.90 (2H, m, CH2-O), 3.65 (1H, t, CH2-NCO), 3.48 (1H, t, CH2-NCO), 3.42 (1H, t, CH2-NCO), 3.19 (1H, t, CH2-NCO), 2.68 (2H, t, CH2-N), 2.42 (2H, m, CH2-O), 1.68 (8H, m, CH2), 1.31 (18H, m, CH2), 0.90 (3H, t, CH3). 1H-NMR (DMSO-d6, at 75°C) δ 3.90 (2H, t, CH2-O), 3.65 (2H, t, CH2-NCO), 3.47 (2H, t, CH2-NCO), 2.74 (2H, t, CH2-N), 2.46 (2H, t, CH2-CO), 1.90-1.33 (26H, m, CH2), 0.90 (3H, t, CH3).
3. Spectral Analysis
- The analysis of the 1H-NMR spectrum for compound 1, which can also be applied to compound 2 is reported in this analysis. As mentioned in the abstract, compounds 1 and 2 are prepared as two intermediates during the synthesis of the natural product acarnidine 3 and its analog 3a1. The 1H-NMR spectra were determined with a Bruker AC-E 300 spectrometer using CDCl3, DMSO-d6, or pyridine-d5 as solvents. The full 1H-NMR spectrum of 1 with assignments in CDCl3 at 20°C and part of that spectrum in the 2-4 ppm region with assignments in DMSO-d6 at 20°C and at 75°C are shown in Figure 1. In both CDCl3 and DMSO-d6 at 20°C, the spectra indicated that two rotamers, 1a and 1b, are present while in DMSO-d6 or in pyridine-d5 at 75°C the spectrum reduced to its simple form because now the two rotamers equilibrate too fast for the NMR instrument to be able to differentiate between them. In DMSO-d6 at 20°C, four apparent triplets (f, f, f, f) at 3.2 to 3.6 ppm were observed which coalesced at 75°C to two triplets (f, f) at 3.4 and 3.5 ppm. Also at 75°C, the apparent multiplets for d, e, and h protons were reduced to the simple triplets at 2.4, 2.7, and 3.8 ppm respectively. In CDCl3 at 20°C, d and h appeared as two multiplets, e as two multiplets, and f and f as three apparent multiplets instead of the four apparent triplets as indicated in pyridine-d5 at 20°C. Three apparent triplets appeared because two of the f signals undergo superposition on each other. The simple spectrum cannot be seen in CDCl3 since it is not feasible to heat this solvent to the required higher temperature. To summarize, at lower temperature the two rotamers are present and detected by the 1H-NMR instrument due to the contribution of the hydrogen bonding. At higher temperature the contribution of the H-Bonding is diminished to become unnoticeable at 75°C as the spectrum in DMSO-d6 demonstrated. One can conclude that the two rotamers 1a and 1b can be separated at low temperature [5].
4. Discussion and Conclusions
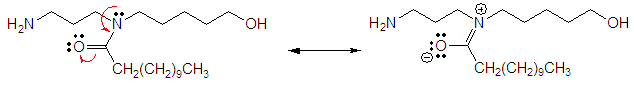
- As we know amides are known to exhibit complex spectra because of the non-equivalence of the two rotamers. Thus, the methyl groups of dimethyformamide (not shown here) may appear as two separate singlets because of the slow inversion process. For this work’s spectral analysis, the two-amide rotamers, 1a & 1b, were present and detected by the 1H-NMR instrument; their presence is mainly due to hydrogen bonding of the rotamers and the solvents (Figure 2) and among themselves as shown in Figure 3. As the hydrogen bonds diminish, the rotamers would equilibrate faster, and hence, the 1H-NMR spectrum appears in its simplest form. The polar aprotic solvents used such as DMSO and pyridine allow the formation of such a bond with certain extent. Hydrogen bond diminishes at higher temperature as demonstrated by the 1H-NMR spectrum in DMSO-d6 at 75°C where the simple spectrum of compound 1 with no rotamers is shown. In DMSO-d6 at 20°C the hydrogen bonds do exist and therefore the complexed spectrum of the rotamers present. Also this complexed spectrum is demonstrated in CDCl3 at room temperature and due to solvent boiling point the simplified spectrum could not be achieved since it is not feasible to elevate the temperature higher. Another factor, besides hydrogen bonding, that hinders the rotation hence allowing the appearance of two rotamers is the presence of a lone pairs of electrons around the central nitrogen atom. The lone pair of the nitrogen amid allows resonance with the carbonyl group and an apparent of double bond forms as shown below. As we know presence of double bonds restrict rotations around them which allows the formation of two conformers.
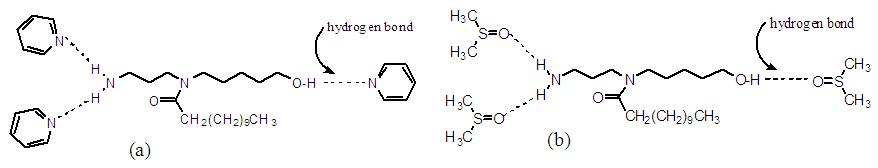 | Figure 2. (a) Hydrogen bounding between compound (1) and the solvent pyridine. (b) Hydrogen bounding between compound (1) and the solvent DMSO |
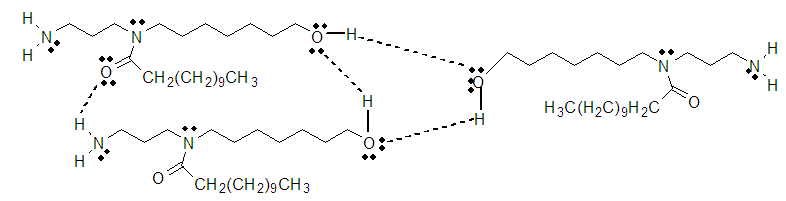 | Figure 3. Hydrogen bounding among molecules of rotamers (compound 1) |
|
|