-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Virology and Molecular Biology
p-ISSN: 2163-2219 e-ISSN: 2163-2227
2025; 14(3): 35-42
doi:10.5923/j.ijvmb.20251403.03
Received: May 26, 2025; Accepted: Jun. 13, 2025; Published: Jun. 17, 2025

Thermodynamic Analysis of the Reactions of the Sulfonation Process of C7-C18 Alkylbenzenes with Sulfuric Anhydride
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLTurdikulov Tolmas Nurulloyevich1, Doniyarov Giyas Tilavovich2, Bahramov Hasan Kayumovich3, Kadyrov Hassan Irgashevich4
1Base Doctoral Student, Tashkent Chemical-Technological Institute, Department of "Basic Organic Synthesis", Tashkent, Republic of Uzbekistan
2PhD., Tashkent Chemical-Technological Institute Yangiyer Branch, Yangiyer City, Republic of Uzbekistan
3PhD, Bukhara State Medical Institute, Head of the Department of "Medical Chemistry", Bukhara, Republic of Uzbekistan
4Doctor of Technical Sciences, Professor of the department of Basic Organic Synthesis, Tashkent Institute of Chemical Technology, Tashkent, Republic of Uzbekistan
Correspondence to: Turdikulov Tolmas Nurulloyevich, Base Doctoral Student, Tashkent Chemical-Technological Institute, Department of "Basic Organic Synthesis", Tashkent, Republic of Uzbekistan.
| Email: |  |
Copyright © 2025 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

This study presents a comprehensive thermodynamic analysis of the sulfonation reactions of linear, isostructured, and unsaturated C7–C18 alkylbenzenes with sulfur trioxide (SO₃), aimed at optimizing the production of alkylbenzene sulfonic acids (ABSA)—precursors for surfactant synthesis. Utilizing the DFT method in the Gauss computational package, standard thermodynamic parameters (ΔG, ΔH, ΔS) for various target and side reactions were calculated. Results demonstrated that the sulfonation of alkylbenzenes is thermodynamically favorable across all studied substituent lengths, with Gibbs free energy values ranging from approximately –185 to –235 kJ/mol depending on the molecular structure. It was found that para-substitution is preferred due to steric and electronic effects, and the formation of by-products such as pyrosulfonic acids and sulfones is also feasible under industrial conditions. A mathematical model of the sulfonation reactor was proposed, integrating mass and heat transfer dynamics as well as operational cycles. The model confirmed that optimizing process parameters enables nearly complete conversion of raw materials while mitigating high-viscosity by-product formation. These findings support the development of more efficient and sustainable industrial sulfonation processes for surfactant production.
Keywords: Pyrolysis distillate, Fractional distillation, Arenes, Sulfonation, Alkanolamines, Ammonium salts, Surfactants
Cite this paper: Turdikulov Tolmas Nurulloyevich, Doniyarov Giyas Tilavovich, Bahramov Hasan Kayumovich, Kadyrov Hassan Irgashevich, Thermodynamic Analysis of the Reactions of the Sulfonation Process of C7-C18 Alkylbenzenes with Sulfuric Anhydride, International Journal of Virology and Molecular Biology, Vol. 14 No. 3, 2025, pp. 35-42. doi: 10.5923/j.ijvmb.20251403.03.
1. Introduction
- In many countries, the deep processing of products formed during the pyrolysis process has also been established, in which various valuable reagents are obtained. For example, in this article, the authors studied the yield of naphthalene by thermal treatment of heavy pyrolysis resin. In the work, distillate fractions and cubic residue were obtained by atmospheric-vacuum distillation by treating heavy pyrolysis resin in a reactor with a stirrer at a temperature range of 250-270°C for 6-8 hours. The distillate was separated by distillation into fractions in a short temperature range: main fraction (up to 200°C), naphthalene fraction (200 - 230°C), methylnaphthalene fraction (230 - 245°C), residual fraction (245 - 340°C).As a result of the thermal treatment carried out by the authors, an increase in the naphthalene fraction and the content of naphthalene in it was observed, while a decrease in the content of mono- and bicyclic alkenes and dienes, vinyl aromatic hydrocarbons, indene and its homologues, dihydronaphthalenes in the product was observed [1].Oil-polymer resins with targeted complex properties, used as a functional additive to polymer compositions with a high yield under certain conditions of thermal processing of heavy pyrolysis resins, as well as commercially pure naphthalene, can be isolated without expensive additional processing.To date, known methods for utilizing liquid pyrolysis products are mainly aimed at isolating technical hydrogenation products of certain fractions, as well as individual products (benzene, toluene, xylene, dicylopentadiene, naphthalene, etc.) for further use. In addition, liquid pyrolysis products are also utilized in the direction of producing low molecular weight "oil polymer resins" [2].
2. Materials and Methods
- Study Design and ParticipantsThis study employed a computational and theoretical design to simulate the sulfonation reactions of C7–C18 alkylbenzenes under industrial conditions. Conducted during 2024–2025, the research did not involve human or biological participants but focused on modeling molecular behavior through thermodynamic and quantum-chemical approaches. No sampling or sample size calculations were required in the classical empirical sense. The study considered various classes of alkylbenzenes—linear, isostructured, and unsaturated—across different carbon chain lengths (C9–C14) for comparative analysis. The selection criteria for these molecules were based on industrial relevance, availability in pyrolysis products, and variation in side-chain structure, while no specific exclusion criteria were applied due to the theoretical nature of the study.Data Collection InstrumentsThe main instruments utilized for data collection and computational modeling were advanced quantum-chemical simulation tools. Thermodynamic properties—Gibbs free energy (ΔG), enthalpy (ΔH), and entropy (ΔS)—were calculated using the Gaussian (Gauss) software package based on Density Functional Theory (DFT). The simulation relied on parameterized molecular descriptors, and geometry optimization was conducted to ensure the stability of molecular structures. The DFT method was selected due to its balance between accuracy and computational efficiency. Validity of the data was ensured through cross-validation with known literature values, and reliability was supported by repeated calculations under varying conditions, all yielding consistent thermodynamic trends.ProceduresThe research procedure did not require participant access or questionnaire-based settings. Instead, alkylbenzene molecules were designed and structurally optimized using computational chemistry environments. The setting was a controlled digital laboratory involving energy minimization routines and quantum mechanical calculations. Raw data in the form of molecular energies and spatial configurations were input into the Gauss software, which performed the DFT-based simulations. All computational runs were performed using standardized reaction conditions (303 K, 110 kPa) mimicking industrial reactor environments. The generated outputs, including reaction energies and equilibrium conditions, were collected and tabulated for analysis. No physical materials or chemical reagents were handled during the study.Data AnalysisAll data analyses were performed using the Gauss software suite and supplemented with Excel for statistical tabulation and graph generation. Thermodynamic parameters were derived by calculating energy differences between reactants and products, as well as transition states where applicable. The DFT method was specifically chosen for its effectiveness in evaluating electron density distributions and predicting thermochemical behavior. Multiple reaction pathways, including para-substitution, sulfonation into side chains, and by-product formation (e.g., sulfones, pyrosulfonic acids), were analyzed. The justification for using DFT lies in its widespread acceptance for modeling chemical reactivity and its ability to incorporate electron correlation effects. Summary statistics (mean, range) were computed across different molecule classes to assess consistency and reaction favorability.
3. Results
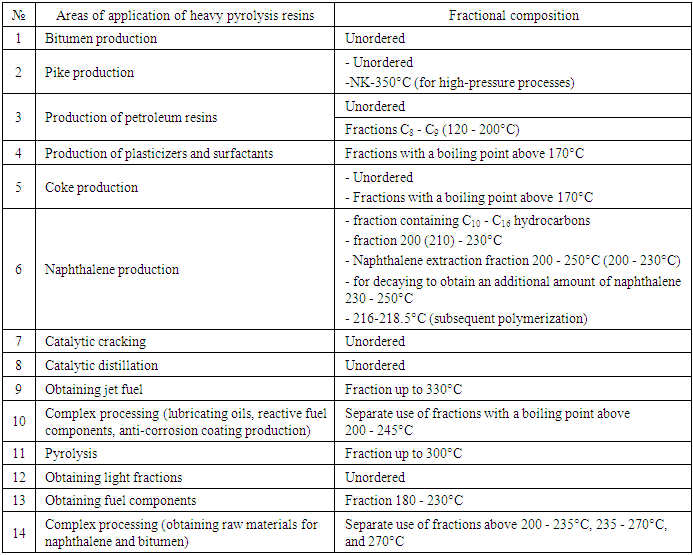
- Foreign companies currently produce 30-40 types of petroleum polymer resins of various nature and applications [3], and in our country, the products of the pyrolysis process are still used as fuel.Heavy pyrolysis resin is a liquid secondary product of the pyrolysis process. It contains aromatic compounds C8+, resins, asphaltenes, which are formed as a result of the condensation and condensation reaction of aromatic rings [4]. It has been established that the content of naphthalene and methylnaphthalene exceeds 25% [5,6]. In modern pyrolysis plants, heavy pyrolysis resin is not considered the main product, it does not have a stable composition, physicochemical properties, which change depending on the composition of the initial raw material and the technological regime. The use of heavier raw materials for the pyrolysis process leads to an increase in the content of naphthalene and its homologues (more than 25%) in the heavy pyrolysis resin [7]. The modern use of heavy pyrolysis resin has quite broad directions, but it has not solved the problem of its full utilization (1-table).It is known that the chemical composition of heavy pyrolysis resin is complex and in many cases, it is impossible to identify the composition under production conditions. Heavy pyrolysis resin is a by-product of the process, its analysis methods are expensive and not always feasible, therefore it is mainly used as a raw material component in various processes (1-table).
|
|
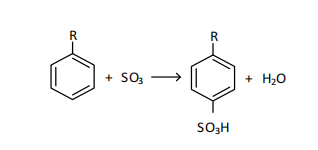 where R - is a linear hydrocarbon substituent with a chain length of 9-14 carbon atoms.
where R - is a linear hydrocarbon substituent with a chain length of 9-14 carbon atoms.
|
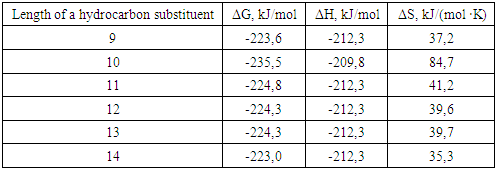
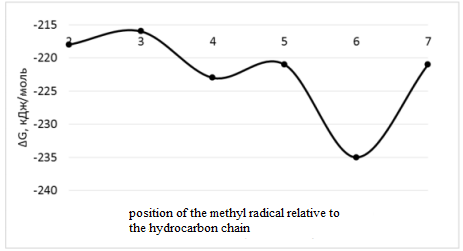 | Figure 1. Dependence of the Gibbs energy value on the position of the methyl radical relative to the hydrocarbon chain |
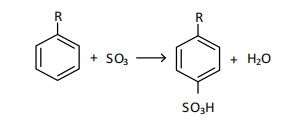 where R- is an isostructured hydrocarbon substituent with a chain length of 9-14 carbon atoms
where R- is an isostructured hydrocarbon substituent with a chain length of 9-14 carbon atoms
|
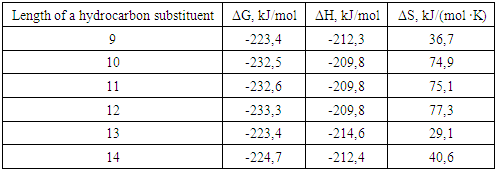
 where R - is an unsaturated hydrocarbon with a chain length of 9-14 carbon atoms, at the sixth carbon atom.
where R - is an unsaturated hydrocarbon with a chain length of 9-14 carbon atoms, at the sixth carbon atom.
|
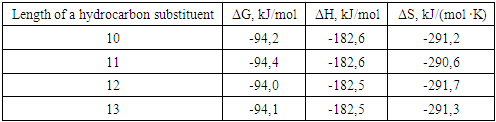
 where R - is a hydrocarbon substituent containing an unsaturated structural double bond with a chain length of 10-13 carbon atoms, located on the sixth carbon atom, SO3 - on the sixth carbon atom.
where R - is a hydrocarbon substituent containing an unsaturated structural double bond with a chain length of 10-13 carbon atoms, located on the sixth carbon atom, SO3 - on the sixth carbon atom.
|
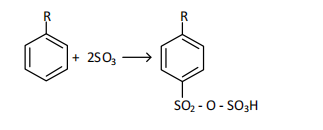 where R- is an unsaturated structural hydrocarbon substituent with a chain length of 10-13 carbon atoms.
where R- is an unsaturated structural hydrocarbon substituent with a chain length of 10-13 carbon atoms.
|
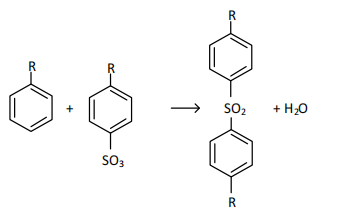 where R - is an unsaturated structural hydrocarbon substituent with a chain length of 10-13 carbon atoms.
where R - is an unsaturated structural hydrocarbon substituent with a chain length of 10-13 carbon atoms.
|
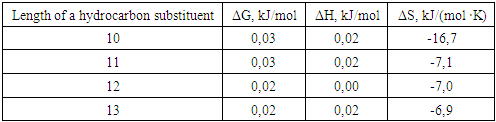
4. Discussion
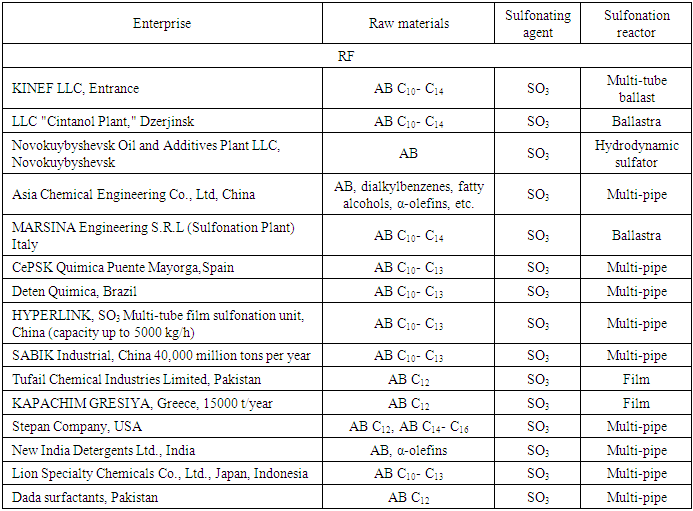
- The thermodynamic modeling conducted in this study has provided valuable insights into the sulfonation behavior of C7–C18 alkylbenzenes under industrial conditions. The use of quantum-chemical simulations based on Density Functional Theory (DFT) has proven highly effective for evaluating the feasibility, stability, and reaction pathways of these compounds, which are essential intermediates in the production of surfactants such as alkylbenzene sulfonic acids (ABSA).A key finding is the relative independence of sulfonation thermodynamics from the length of the hydrocarbon side chain. Despite variations in chain length (C9 to C14), the Gibbs free energy (ΔG) for the sulfonation reactions remained consistently within a narrow range, averaging around –225 to –228 kJ/mol for linear and isostructured alkylbenzenes. This indicates a broadly favorable thermodynamic profile across this class of compounds and suggests high adaptability in the choice of raw materials for industrial ABSA production.An important implication of this result is the feasibility of unifying the industrial sulfonation process for a broader range of feedstocks, including unsaturated and branched alkylbenzenes, without substantial alterations in reaction conditions. Notably, even the sulfonation of unsaturated alkylbenzenes showed only a moderate thermodynamic deviation, supporting their inclusion in process streams. However, the slightly reduced ΔG for side-chain sulfonation compared to para-substitution underscores the preference for the latter due to steric and electronic stabilization, aligning with previous experimental data on electrophilic aromatic substitution.The study also confirms the thermodynamic plausibility of side reactions such as the formation of sulfones and pyrosulfonic acids. These by-products, while less favorable than the main sulfonation pathways, can accumulate under certain process conditions and contribute to high-viscosity residues that impair reactor hydrodynamics. The close-to-zero Gibbs energy observed for sulfone formation highlights the potential equilibrium nature of such reactions, reinforcing the necessity of precise process control to minimize their occurrence.Furthermore, the modeling of reactor behavior through activity coefficients and the inclusion of heat and mass transfer variables allowed the development of a refined mathematical model of the sulfonation reactor. This approach not only quantifies the conversion efficiency but also offers practical guidance for adjusting operational parameters such as washing intervals and raw material throughput, thus enabling nearly complete conversion of feedstocks while suppressing undesirable side reactions.Comparing these findings with global industrial practices—such as those of KINEF (Russia), Stepan Company (USA), and MARSINA (Italy)—it is evident that the adoption of multi-tube film reactors and accurate raw material metering, as modeled in this study, are critical for optimizing yields and ensuring sustainability. The study provides scientific validation for current technological trends and lays a theoretical foundation for their further refinement.This thermodynamic analysis confirms the high efficiency and robustness of the industrial sulfonation of alkylbenzenes. The minimal influence of side chain variations on the overall thermodynamic favorability supports the expansion of raw material diversity, while the accurate prediction of by-product formation offers routes to better reactor design and process optimization. These findings have practical significance for surfactant manufacturing industries aiming to enhance conversion rates, reduce waste, and maintain product quality under varying feedstock conditions.
5. Conclusions
- This study has demonstrated that the sulfonation of C7–C18 alkylbenzenes with sulfur trioxide is thermodynamically favorable across a wide range of molecular structures, including linear, isostructured, and unsaturated variants. The application of quantum-chemical methods, particularly Density Functional Theory (DFT) using the Gauss software package, allowed for accurate calculation of key thermodynamic parameters (ΔG, ΔH, ΔS), which consistently indicated strong reaction feasibility.The results confirm that para-substitution is the most energetically preferred sulfonation pathway, primarily due to favorable electronic and steric effects. While side-chain sulfonation and by-product formation (e.g., pyrosulfonic acids and sulfones) are also thermodynamically possible, their occurrence can be minimized through precise control of reactor conditions. The near-constant Gibbs energy values across different alkyl chain lengths support the standardization of industrial processes, enabling flexibility in raw material selection without compromising reaction efficiency.Moreover, the developed mathematical model of the sulfonation reactor provides a practical tool for optimizing operational parameters, improving conversion rates, and reducing the accumulation of high-viscosity by-products. Overall, this work offers both theoretical insight and practical value for enhancing the sustainability and performance of industrial surfactant production systems.