-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Virology and Molecular Biology
p-ISSN: 2163-2219 e-ISSN: 2163-2227
2020; 9(2): 17-34
doi:10.5923/j.ijvmb.20200902.01

High Risk Functional nsSNP in SARS-CoV-2 (2019-nCoV) Main Peptidase as Potential Targets to Structure-Based Drug Design: A Computational Approach
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLSahar G. Elbager 1, Abdelrahman H. Abdelmoneim 2, Afra M. Al Bkrye 3, Asia M. Elrashied 4, Entisar N. M. Ali 5, Hadeel A. Mohamed 6, Hazem A. Abubaker 7, Israa A. Mohamed 8, Manal A. H. Goda 9, Mohammed Y. Basher 7, Naglla F. A. Gabir 4, Safinaz I. Khalil 10
1Faculty of Medical Laboratory Sciences, University of Medical Sciences and Technology (UMST), Khartoum, Sudan
2Faculty of Medicine, Alneelain University, Khartoum, Sudan
3College of Veterinary Medicine, University of Bahri, Khartoum, Sudan
4Faculty of Science, University of Khartoum, Khartoum, Sudan
5Faculty of Medical Laboratory, University of Kordofan, Kordofan, Sudan
6Faculty of Science and Technology, Omdurman Islamic University, Sudan
7Faculty of Veterinary Medicine, University of Khartoum, Khartoum, Sudan
8Faculty of Science and Technology, University of Bahri, Khartoum, Sudan
9Institute of Endemic Diseases, Khartoum, Sudan
10Faculty of Medicine, Al Fajr College for Science and Technology, Khartoum, Sudan
Correspondence to: Sahar G. Elbager , Faculty of Medical Laboratory Sciences, University of Medical Sciences and Technology (UMST), Khartoum, Sudan.
| Email: |  |
Copyright © 2020 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

On January 2020, a new coronavirus (officially named SARS-CoV-2) was associated with alarming outbreak of a pneumonia-like illness, which was later named by the WHO as COVID-19, originating from Wuhan City, China. Although many clinical studies involving antiviral and immunomodulatory drug treatments for COVID-19, no approved drugs have been found to effectively inhibit the virus so far. A promising target for SARS-COV-2 drug design is a (Mpro), a main protease (Mpro), responsible for the replication and maturation of functional proteins in the life cycle of the SARS coronavirus. Here we employed missense SNP analysis to all amino acid identified as functional residues and showed potential binding activities to predict the high risk missense mutations that are highly damaging to the structure and the function of the SARS-COV-2 Mpro aiming to detect potential drug targets for inhibition of SARS-COV-2 Mpro. Our results demonstrated essential roles of HIS41, GLY143, CYS145, HIS163, GLU166 as binding residues, structural and functional residues, participating in different physicochemical interaction for keeping the proteolytic activity and enzymatic function of the SARS-COV-2 Mpro. Upon subjecting these residues to single-nucleotide polymorphism (SNP) analysis, out of 152 nsSNP, 34 nsSNP recognized to alters the functionality and stability of SARS-COV-2 Mpro. Furthermore, most of these deleterious nsSNPs were predicted as ligand binding residues. We conclude that these residues can be target sites for the fresh generation inhibitors for of the SARS‐CoV Mpro and overcome the current drugs ineffectiveness.
Keywords: COVID-19, Coronaviruses, SARS-COV-2, Main Protease, Mpro
Cite this paper: Sahar G. Elbager , Abdelrahman H. Abdelmoneim , Afra M. Al Bkrye , Asia M. Elrashied , Entisar N. M. Ali , Hadeel A. Mohamed , Hazem A. Abubaker , Israa A. Mohamed , Manal A. H. Goda , Mohammed Y. Basher , Naglla F. A. Gabir , Safinaz I. Khalil , High Risk Functional nsSNP in SARS-CoV-2 (2019-nCoV) Main Peptidase as Potential Targets to Structure-Based Drug Design: A Computational Approach, International Journal of Virology and Molecular Biology, Vol. 9 No. 2, 2020, pp. 17-34. doi: 10.5923/j.ijvmb.20200902.01.
Article Outline
1. Introduction
- The innovative SARS-COV-2 in hominids, first revealed in Wuhan, China, in December 2019. The first cases were classified as "pneumonia of unknown etiology" as they were unable to identify the causative agent [1]. The genotyping analysis and phylogenetic relationships of viruses isolates, displays that the viruses belong to genera Human Betacoronavirus (SARS-CoV, and MERS-CoV) have many similarities, but also have differences in their genomic and phenotypic structure that can influence their pathogenesis [2-4]. Subsequently, the International Committee on Taxonomy of Viruses (ICTV) termed SARS-COV-2 (SARS-COV-2 virus) and confirmed as the causative agent of disease (5). On 30 January the World Health Organization has declared this a pandemic worldwide (6). However, there is a lack of specific antiviral treatment recommended for COVID-19, and no vaccine is currently available.Human SARS-COV-2 are enveloped viruses causes a severe acute respiratory tract infection with a high fatality rate in human. SARS-COV-2 are enveloped viruses possesses a ~26.4–31.7 kb positive RNA genome associated with a nucleoprotein within a capsid comprised of matrix protein. The betacoronavirus genome encodes several structural proteins, including glycosylated spike (S), envelope (E), membrane protein (M), and nucleoprotein (N). In addition, the viral genome also encodes tow overlapping polypeptides open reading frame (pp1a and pp1ab) that encode for the replicase [7,8]. Replication of SARS-COV-2 is achieved by translated polypeptides open reading frame (pp1a and pp1ab), which further then cleaved by main peptidase (Mpro) into 16 functional polypeptides (non-structural proteins or nsp) that mediated replication and transcription of the viral genome [9,10]. The SARS-COV-2 Mpro, also known as 3C-like protease (3CLpro or Nsp5), like other coronaviruses, is 306 amino acids long with a molecular weight of 3797.0 Da. This enzyme naturally forms a dimer each monomer consists of three domains. Domain I (residue 8–101) Domain II (residue 102–184) and Domain III (residue 201–303) involves of 5 α-helices (α5-α9), which are connected by a long loop (residues 185-200) with domain II. Domain I and II are important functional domains that includes conserved His41 and Cys145 catalytic dyad, form a substrate binding region. Domain III mediating the tight dimerization of the enzyme, as the protease is active only in dimeric conformation [11,12]. In addition to the catalytic centre there are five subsites (S1-S5). S1& S2 subsites are deeply buried, mainly involved in hydrophobic and electrostatic interactions while S3-S5 subsites, are superficial, involved in different functionalities. The S1 subsite consists of His163, Glu166, Cys145, Gly143, His172, and Phe140 while S2 consist of Cys145, His41, and Thr25 amino acid residues. The superficial subsites S3-S5 comprises Met49, His41, Met165, Glu166 and Gln189 amino acid residues [13-15]. (Figure 1).
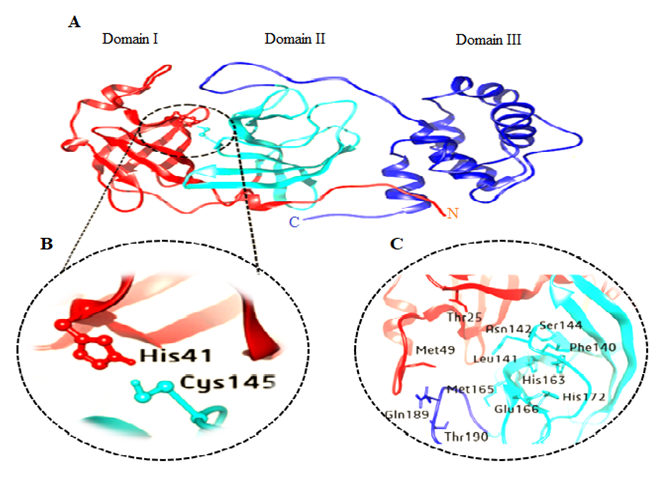 | Figure 1. A) Cartoon representation of 3D structure of one monomer of the dimeric SARS-COV-2 Mpro B) His41- Cys145 catalytic dyad. C) substrate-binding residues within S1-S5 |
2. Material and Method
2.1. Protein Sequence Analysis
- The protein sequence of SARS-COV-2 (Mpro) (accession ID: YP_009725301) was retrieved from the NCBI Protein database (https://www.ncbi.nlm.nih.gov/protein/) on 10 April 102020. In order to identify similar sequences, Sequence comparison and alignment of SARS-COV-2 Mpro were performed using BLASTp (http://blast.ncbi.nim.nih.gov/ /Blast.cgi?PAGE=Proteins).
2.2. Protein of 3D Structure Retrieval and Verification
- All the crystal structures of SARS-COV-2 Mpro were retrieved from Protein Data Bank Protein (PDB; http://wwpdb.org). Chimera used for the validation of 3D structure by plotting Ramachandran plot. UCSF Chimera is a highly extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, and conformational analysis Chimera (version 1.8) [17].The Ramachandran plot analysis provides a simple view of the conformation of a protein. The distinct regions in the Ramachandran plot based on φ-ψ angles cluster reflect a particular secondary structure. Residues are shown as blue dots, or when selected, as red dots colors in the plot indicate most favorable, allowed, generously allowed and disallowed regions. Probability contours based on a reference set of high-resolution proteins can be shown on the plot as green lines. Ideally, a model having more than 90% residues in favourable region is considered as a good-quality protein structure.
2.3. Prediction of Ligand Binding Sites
- Ligand binding sites predictions in SARS-COV-2 Mpro were analyzed using COACH. COACH is a meta-server approach for the prediction for ligand binding targets through two comparative methods TM-SITE and S-SITE [18]. PDB file as an input. The overall results were given in the form of position of potential drug binding sites.
2.4. Identification of Functional Sites (PFSs)
- Active site amino acid residues in SARS-COV-2 Mpro was identified using fiDPD. fiDPD is a sequence-based tool for the prediction of protein function-site and protein-ligand interaction [19]. The method is based on a functional site and physicochemical interaction-annotated domain profile database using protein domains found in the Protein Data Bank. The residues predicted as active residues were subjected to conservation analysis and missense SNP analysis.
2.5. Protein Conservation Analysis
- In order to identify key functional or structural /conserved residues, ConSurf web-server use to calculates the evolutionary conservation of amino acid substitution in proteins [20]. ConSurf gives the output in the form of score where score 9 represent the most conserved and 1 represent the highly variable amino acid.
2.6. Retrieval of Missense Single-Nucleotide Polymorphisms
- All amino acid identified as functional residues and showed potential binding activities, were subjected to missense SNP analysis.
2.7. Prediction of Functional Consequences of nsSNPs
- PROVEAN, PredictSNP1.0, SNPs & GO and Meta-SNP were used to assess the potential functional effect of the missense SNPs. SNPs were classified as Damaging by at least three tools.2.7.1. PROVEAN (Protein Variation Effect Analyzer) is used to predict the possible impact of a substituted amino acid and indels on protein structure and biological function. It analyses the nsSNPs as Damaging or natural, if the final score was below the threshold score of −2.5 were considered Damaging; scores above this threshold were considered neutral [21]. The input query is a protein FASTA sequence along with amino acid substitutions.2.7.2. SNPs & GO web server was used to predict the human disease related single point protein mutations. This server was mainly based on support vector machines which can corroborates all the information regarding variations from the existing databases. It annotates variations as Damaging based on information derived from Gene Ontology (GO) Predictor with overall accuracy of 82% [22]. Protein FASTA sequence along with name and position of wild type and mutant amino acid was submitted as input for this server.2.7.3. PMut a web-based tool for the annotation of pathological variants on proteins. PMut method is based on the use of neural networks (NNs) trained with a large database of neutral mutations (NEMUs) and pathological mutations of mutational hot spots, which are obtained by alanine scanning, massive mutation, and genetically accessible mutations [23]. The final output is displayed as a pathogenicity index ranging from 0 to 1, and the cut-off value is set to 0.5 (neutrual, 0 to 0.5; pathological, 0.5 to 1).2.7.4. PredictSNP1.0 was used as the predictor of the disease related single point protein mutations [24]. PredictSNP is a consensus classifier that integrates the results from nine in silico prediction tools: SIFT, PolyPhen-1, PolyPhen-2, MAPP, PhD-SNP, SNAP, PANTHER, PredictSNP, and nsSNPAnalyzer, thus resulting in significantly improve prediction performance. The input query is a protein FASTA sequence along with amino acid substitutions.2.7.5. Meta-SNP is a random forest-based binary classifier for prediction of disease causing variants to reduce the bias of a single predictor, Meta-SNP was used since it integrates four existing methods: PANTHER, PhD-SNP, SIFT and SNAP [25]. The input query is a protein FASTA sequence along with amino acid substitutions.
2.8. Prediction of Change in Protein Stability due to High Risk nsSNPs
- 2.8.1. I-Mutant 2.0 is a tool used for prediction of changes in protein stability due to single site mutations under different conditions. It is a web server based on support vector machine which worked on dataset derived from Protherm, a database of experimental records on protein mutations [26]. It can predict the stability changes in protein with 80% accuracy based on its structure and with 77% of accuracy based on its sequence. The input can be submitted as either in the form of protein sequence or on a structure basis. 2.8.2. MUpro based on support vector machines and neural networks machine learning methods, which can be used to predict the effects of single-site amino acid mutations on protein stability. MUpro can predict protein stability changes merely using sequence information or combining that information with tertiary structure [27]. A ΔΔG value less than ‘0’ indicates that the variant decreases the protein stability. On the contrary, a ΔΔG value greater than 0 indicates that the variant elevates the protein stability.2.8.3. INPS-MD (Impact of Non-synonymous mutations on Protein Stability-Multi Dimension) is a method used to predict stability of protein variants from sequences and structures. The INPS-MD predictor using sequences is based on a simplified support vector (SVR) as implemented by the libsvm package, which was only tested by linear and radial basis function (RBF) kernels [28]. INPS-MD predictions can be interpreted to identify stabilizing (ΔΔG > 0) and destabilizing (ΔΔG < 0) variations.
2.9. Analyzing the Effect of nsSNPs on 3D Structure of the Proteins and Physiochemical Properties
- Project Hope server was used to search protein 3D structures by collecting structural information from a series of sources, including calculations on the 3D coordinates of the protein, sequence annotations from the UniProt database, and predictions by DAS services [29]. Furthermore, it described the reaction and physiochemical properties of these candidates. Protein sequence and mutant variants were submitted to project hope server in order to analyze the structural and conformational variations that have resulted from single amino acid substitution.
3. Results
3.1. Protein Sequence Analysis
- Sequence comparison and alignment results revealed that SARS-COV-2 Mpro was conserved, with 100% identity among all SARS-COV-2 genomes isolates till April 10, 2020. Then, the SARS-COV-2 Mpro protein sequence was aligned with that of SARS CoV. The results shown that SARS-COV-2 Mpro shares 96.08% sequence identity with SARS-CoV. There were 12 out of 306 residues different between SARS-CoV and SARS-COV-2 (Val35Thr, Ser46Ala, Asn65Ser, Val86Leu, Lys88Arg, Ala94Ser, Phe134His, Asn180Lys, Val202Leu, Ser267Ala, Ala 285Thr and Leu286Iso).
 | Figure 2. Sequence alignment between SARS-COV-2 Mpro and SARS-CoV Mpro. Boxes are displaying mutations |
3.2. Protein of 3D Structure Retrieval and Verification
- The PDB contains more than 80 the 3D structure of the CoV-2 Mpro. Ramachandran plot of SARS-COV-2 Mpro protein structure shows (PDB: 5R7Y) is the best structures identity with the query sequence since most of the residues are present in the core region (99.0) and was chosen for further analysis. Fig. 3 Ramachandran plot analysis of (PDB: 5R7Y).
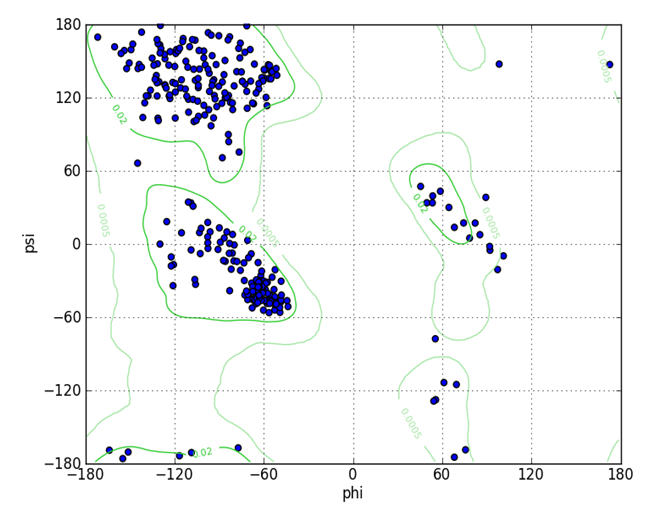 | Figure 3. Ramachandran plot analysis of SARS-COV-2 Mpro protein (5R7Y) showing most of the residues located at the allowed region (the blue dots represent amino acid residues; the green lines indicate the allowed region). (phi) ϕ and (psi) ψ are torsion angles. The torsion angle about the N—C bond is called ϕ and that about the C—C bond is ψ. This analysis is predicted by UCSF Chimera |
3.3. Prediction of Ligand Binding Sites
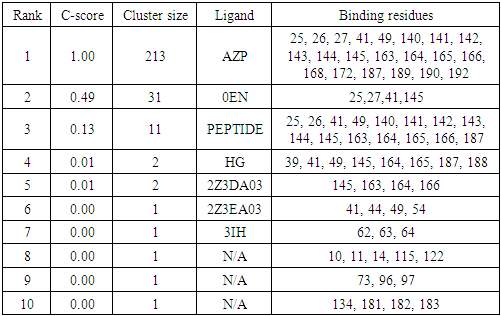
- By using COACH, ligand binding sites in SARS-COV-2 Mpro was analyzed. ligands which have higher C-score (confidence score) indicate a more reliable prediction. The ligand AZP have higher C-score than rest of ligands and its possible binding sites are 25, 26, 27, 41, 49, 140, 141, 142, 143, 144, 145, 163, 164, 165, 166, 168, 172, 187, 189, 190 and 192. Name of ligands and its possible binding sites are shown in Table 1.
|
3.4. Identification of Functional Sites (PFSs)
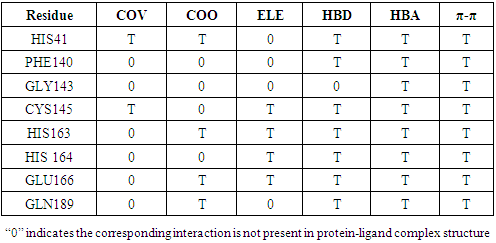
- fiDPD server identified 8 functional sites present in SARS-COV-2 Mpro (HIS41, PHE140, GLY143, CYS145, HIS163, HIS 164, GLU166 and GLN189). Considering the interactions types, fiDPD found very different types of interactions (i.e. covalent bond (COV), coordinate bond (COO), electrostatic interaction (ELE), H-bond donor (HBD), H-bond acceptor (HBA), π-stacking interactions (π-π.). Table 2
|
3.5. Protein Conservation Analysis
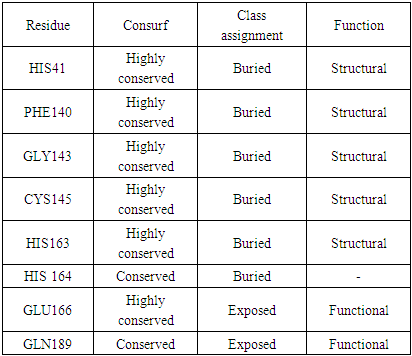
- ConSurf analysis predicted HIS41, PHE140, GLY143, CYS145 and HIS163 to be buried and conserved residue, i.e., a structural residue, which suggests that these positions are important for the SARS-COV-2 Mpro structure. GLU166, GLN189 were predicted to be exposed and conserved residue, i.e., a functional residue, which suggests that these positions are important for the SARS-COV-2 Mpro function. Table 3
|
3.6. Retrieval of Missense Single-Nucleotide Polymorphisms Datasets
- All amino acid residues showed potential functional sites, were subjected to missense SNP analysis. These amino acids include (HIS41, PHE140, GLY143, CYS145, HIS163, HIS164, GLU166 and GLN189), were mutated to all possible missense mutation. SNPs identified theoretically by exchange of each 8 amino acid residues with all possible other nineteen amino acid (8×19 = 152 possible SNPs). Then a variety of computational tools were employed in order to determine the effect of a given missense mutation on the SARS-COV-2 Mpro function and structure.
3.7. Missense Single-Nucleotide Polymorphisms Analysis
- To gather higher accuracy results, five in silico SNP prediction tools (PROVEAN, SNPs&GO, Pmut, PredictSNP and Meta-SNP) were employed to predict the high risk missense SNPs. We categorized SNPs as damaging if they were predicted to be damaging by four or more SNP prediction A total of 152 SNPs were subjected to analysis using theses algorithms. By using PROVEAN tool there were 5 neutrals and 147 Damaging. In SNP & GO there were 90 neutrals and 62 damaging. In PMUT there were 62 neutrals and 90 damaging. In Predict SNP were 9 neutrals and 143 damaging. MetaSNP were 17 neutrals and 135 disease causing. Out of a total of 152 SNPs, 34 SNPs were considered high risk and were subjected to further stability studies.
3.8. Prediction of Change in Protein Stability
- The protein stability change was estimated using I-Mutant 2.0, MUpro and INPS-MD. SNPs were considered as destabilizing if two or more algorithms showed a decrease in stability upon mutation. Out of 34 high risk nsSNPs I-Mutant 3.0 analysis showed that, 25 nsSNPs decreased stability (ΔΔG < 0), whereas 9 nsSNPs increased stability (ΔΔG > 0). MUpro and INPS-MD analysis showed that, 28 and 33 nsSNPs were found to decrease protein stability, respectively. In total Out of 34 high risk nsSNPs, 30 nsSNP were predicted to decrease the stability, 4 nsSNPs were found to increase protein stability. H41L was found to increase protein stability by the three tools.
3.9. Analyzing the Effect of nsSNPs on 3D Structure of the Proteins and Physiochemical Properties
- The common physiochemical consequence of this nsSNPs were alteration in charge, size and hydrophobicity between wild-type and mutant residues which might lead to loss of interactions with other molecules and loss of hydrogen bonds in the core of the protein and as a result disturb correct folding. Table 6 & Fig 4-9.
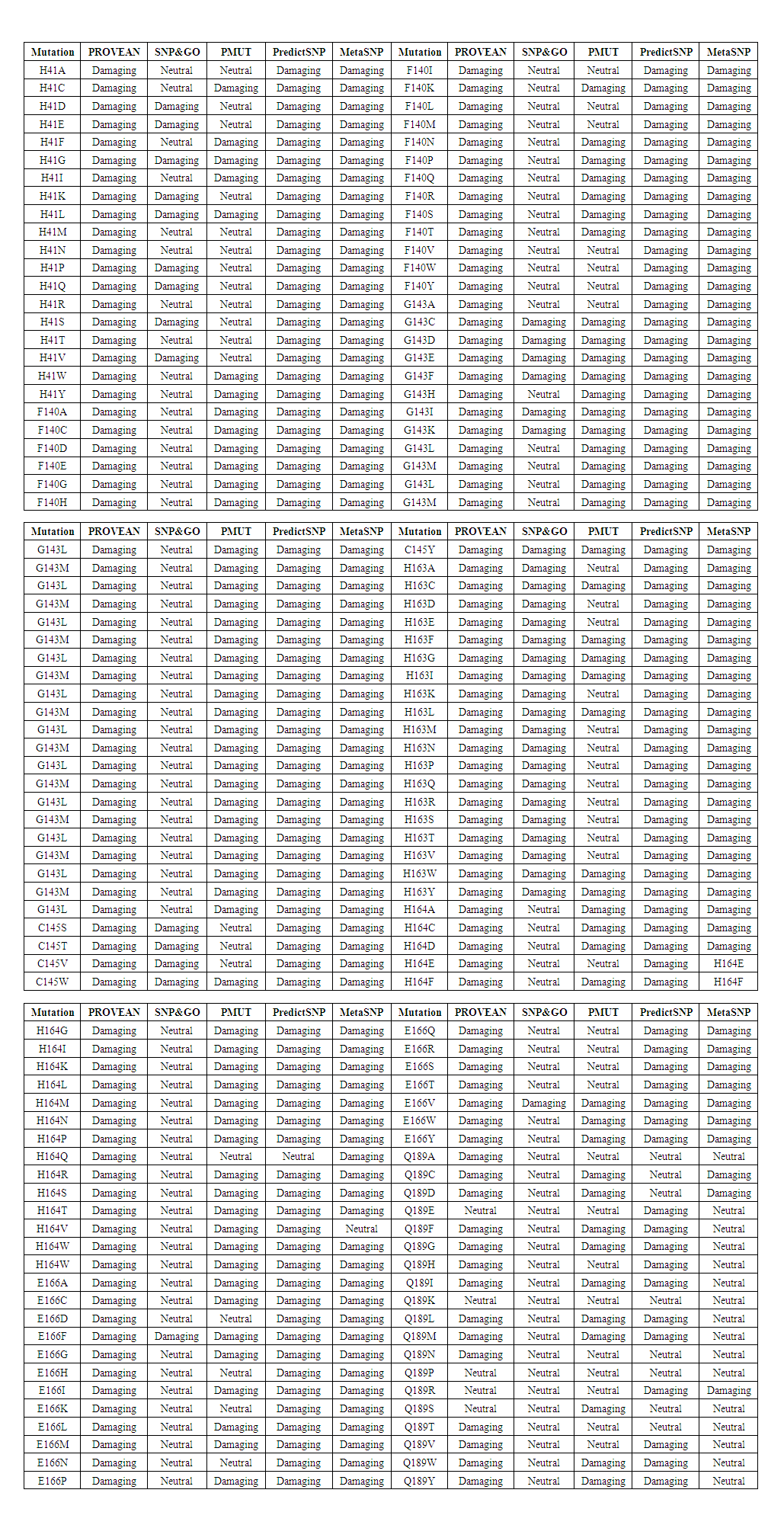 | Table 4. Missense SNPs in Mpro predicted to be damaging/ neutral |
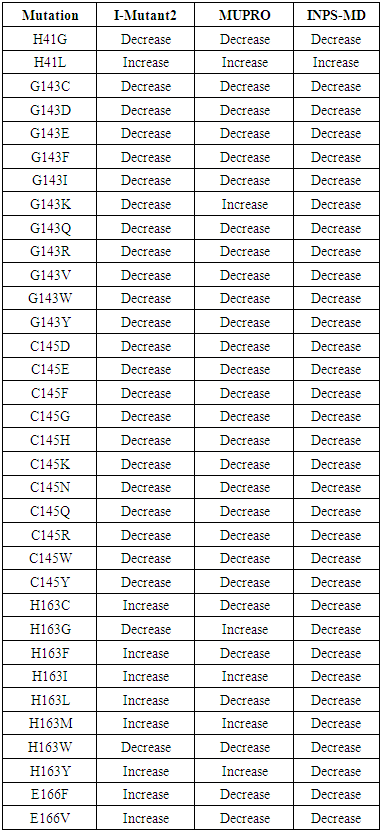 | Table 5. Prediction result of Protein Stability |
 | Table 6. Schematic structures of the wild-type residue and mutant residue amino acid for each Mutation |
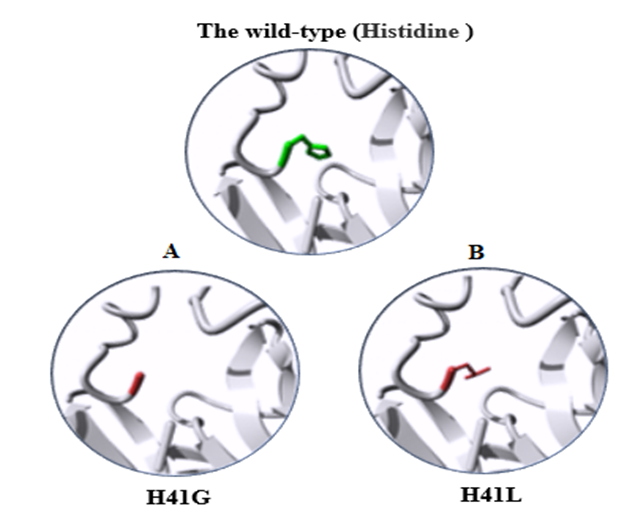 | Figure 4. 3D structure of the wild-type Histidine substitution. A) the mutant residue H41G. B) the mutant residue H41L |
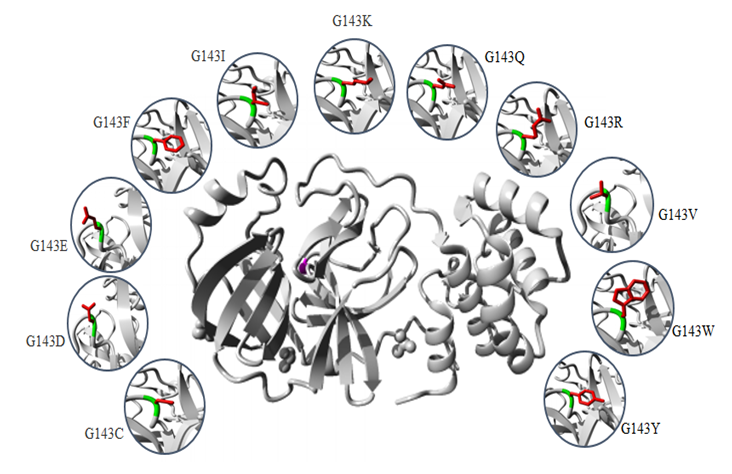 | Figure 5. Structures of the protein (center) is colored grey, the side chain of the mutated residue is colored magenta and shown as small balls. 3D structures of the substitutions of wild-type Glycine at position 143. The wild-type colored green and the mutant residue colored green red |
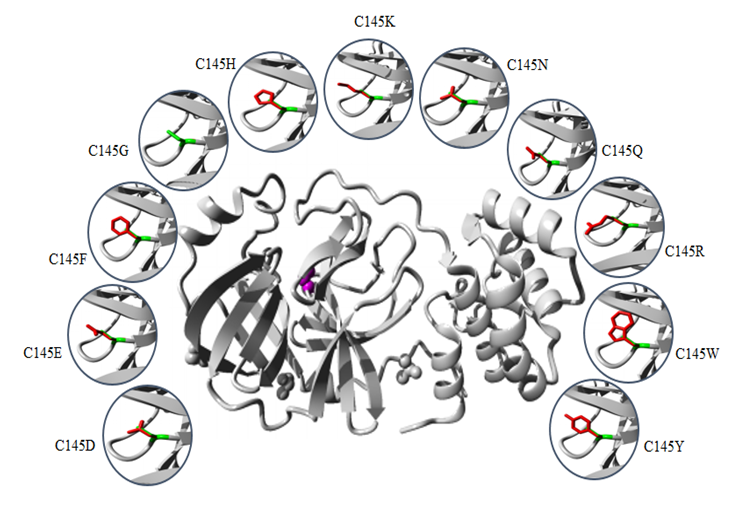 | Figure 6. Structures of the protein (center) is colored grey, the side chain of the mutated residue is colored magenta and shown as small balls. 3D structures of the substitutions of wild-type Cysteine at position 145. The wild-type colored green and the mutant residue colored green red |
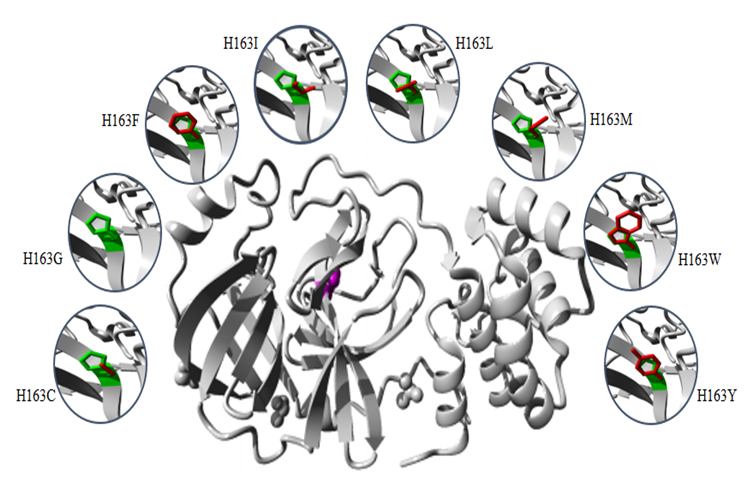 | Figure 7. Structures of the protein (center) is colored grey, the side chain of the mutated residue is colored magenta and shown as small balls. 3D structures of the substitutions of wild-type Histidine at position 163. The wild-type colored green and the mutant residue colored green red |
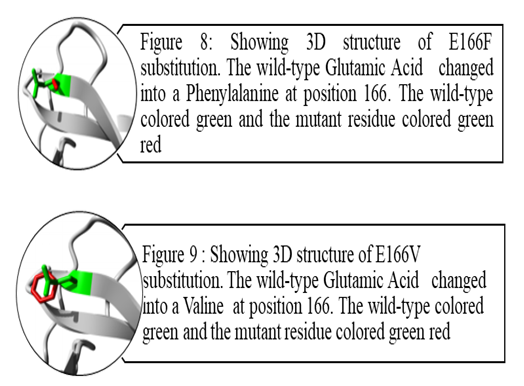 | Figure 8&9 |
4. Discussion
- The SARS-COV-2 Mpro play critical role in the maturation and posttranslational processing of the viral replicase protein and thus propagation of the SARS-COV-2 infections. Therefore, great effort has been spent on studying this protein in order to identify therapeutics against the SARS-COV-2 based on some of the previous progress of specific inhibitors development for the SARS-CoV enzyme because they share similar conserved regions, active sites and enzymatic mechanisms [30-33]. Our study displayed that Mpro is conserved in all SARS-COV-2. It is highly similar to SARS-CoV Mpro, with only 12 residues different. These differences may affect Mpro structure and function and might disrupt important hydrogen bonds and alter the receptor binding site, thereby affecting its ability to bind with the SARS-CoV inhibitor. In the structure-based drug design process, identification of ligand-binding sites in the target protein is the first step. In the literature, several crystal structures of SARS-COV-2 Mpro in complex with different inhibitor are deposited in the protein data bank. In order to predict the ligand-binding in CoV-2 Mpro, firstly the Protein Data Bank (PDB) structures were retrieved and verified. Our analysis shows (PDB: 5R7Y) is the best structures identity with the CoV-2 Mpro sequence and was chosen for ligand-binding sites identification. Ten potential binding sites were predicted using COACH. The ligand AZP have higher C-score possesses 21 binding residues (25, 26, 27, 41, 49, 140, 141, 142, 143, 144, 145, 163, 164, 165, 166, 168, 172, 187, 189, 190 and 192). 8 Out of this residue were identified as functional residues (HIS41, PHE140, GLY143, CYS145, HIS163, HIS 164, GLU166 and GLN189) participating in various physicochemical interaction at the active site of Mpro. particularly GLY145, HIS163, HIS 164 and GLU166 contribute significantly to strong electrostatic interactions in the active site. All these residues were subjected to conservation and missense SNP analysis. Single-nucleotide polymorphisms (SNPs) in protein functional sites may causing loss of protein–ligand interactions, disturb protein dynamics by changing protein stability, disrupting and blocking the active sit resulting in a loss of inhibitor efficiency. The catalytic dyad of Mpro, located at the cleft of domain I and II, includes the conserved residues HIS41 and CYS145, which are essential for catalytic activity. GLY143, HIS163, Glu166 residues are located at domain II and involved in the deeply buried subsite (S1), this domain is important for binding of substrate binding. Mutation of this residues might change the 3D conformation of the substrate-binding subsite and disturb the interaction and as such obstruct substrate binding. Except for PHE140, HIS164 and GLN189, all rest of residues showed to be highly risk damaging SNPs.HIS41 involved in the catalytic centre of SARS-CoV Mpro, play a part in various physicochemical interaction. Based on conservation analysis, HIS41 is a highly conserved and buried in the core of domain I, thus mutation in the HIS41 residue is probably disrupting structure of this domain. The current study predicted that H41G and H41L are highly damaging SNPs destabilizing protein stability. H41 G and H41L resulted in a change of the wild-type histidine at position 41 to glycine and leucine respectively. The mutant residues are smaller and more hydrophobic than the histidine, these differences might disturb the conformation structure of this domain leading to loss of the proteolytic activity of the SARS-COV-2 Mpro. Table 6 & Fig. 4.The GLY143 is located at the buried the oxyanion-binding loop involved in the deeply buried subsite (S1) of Domain II, has an electrostatic interactions and hydrogen bonds with other residues. ConSurf analysis showed that, GLY143 is an important residue for the SARS-COV-2 Mpro structure. Glycines is most flexible of all amino acids residues. This flexibility might be necessary for the protein's function. Any mutation in the GLY143 residue would disturb the required flexibility at the oxyanion-binding loop and therefore disturb a special backbone conformation which might be required at this position. Results of the current study predicted 11 nsSNPs are highly damaging result in the decrease of the protein stability. These mutations introduce some amino acids with different size, hydrophobicity and charge, thus these mutations are probably disrupting structure of this domain and decrease the enzymatic function of the SARS-COV-2 Mpro. Table 6 & Fig. 5.The CYS145 involved in the catalytic center of SARS-CoV Mpro, participating in different physicochemical interaction. Based on conservation analysis, CYS145 is a highly conserved and buried in the core of domain II. Being important structural residue, any substitution in the CYS145 residue would disturb the local structure of domain II. Missense SNP analysis predicted 11 nsSNPs are highly damaging result in the decrease of the protein stability. These mutations introduce some amino acids with different size, hydrophobicity and charge, thus these mutations might cause loss of hydrophobic interactions with other molecules, disrupting structure of this domain and leading to loss or reduce of the proteolytic activity of the SARS-COV-2 Mpro. Table 6 & Fig. 6.The HIS163 is buried in the core of a domain II, involved in several physicochemical interactions with other residues. Conservation analysis showed that, HIS163 is an important residue for the SARS-COV-2 Mpro structure. Results of this study predicted 8 nsSNPs are highly damaging and destabilizing of the protein. The following project hope summary report shows the structural and physiochemical changes induced by these mutations, mainly as variation in the size, charge and hydrophobicity of the final protein. The wild-type histidine forms a hydrogen bond with: Tyrosine at position 161, thus these mutations might cause loss of hydrogen bonds in the core of the protein and as a result disturb correct folding of the SARS-COV-2 Mpro. Table 6 & Fig. 7.The Glu166 located in the subsite (S1) of Domain II, involved in several physicochemical interactions with other residues. Conservation analysis showed that, Glu166 is an important functional residue; highly conserved and exposed in the surface of domain II, thus mutation in the Glu166 residue is probably disrupting function of this domain. Results of the current study predicted E166F and E166V are highly damaging SNPs decrease the protein stability. These mutations resulted in a change of the wild-type glutamic acid at position 166 to phenylalanine and valine respectively. Phenylalanine is bigger and more hydrophobic than glutamic acid, where valine is smaller and more hydrophobic than glutamic acid. Glutamic acid is negatively charged residue forms a hydrogen bond with histidine at position 172, where phenylalanine and valine residues neutrally charged. These differences in size, hydrophobicity and charge might affect hydrogen bond formation, lead to loss of interactions with other molecules, which can disturb the enzymatic function of the SARS-COV-2 Mpro. Table 6 & Fig. 8-9.Previous mutagenesis study in other CoV Mpro established the importance of His41, Cys145, and Glu166 in the substrate‐binding site for keeping the proteolytic activity and enzymatic function of the SARS‐CoV Mpro [34,35]. Moreover site-directed mutagenesis studies in other CoV Mpro revealed that substitution of either His41, Cys145 or H163 residues resulted in complete loss of proteolytic activity [36,37]. Results of the present study determined that HIS41, CYS145, H163 and E166 are highly damaging missense SNPs decreases protein stability except H41L, H163I, H163M, and H163Y SNPs, which increase the protein stability. Due to the urgent need for effective treatments, use of repurposed existing antiviral drugs approved and successes for treatment of other viral infections such as HIV, SARS-CoV-1 and MERS-CoV is somewhat promising. Based on literature search, (HIV-1) protease inhibitors (lopinavir/ritonavir) and new nucleoside analogues (remdesivir) are now most commonly protease inhibitor proposed for potential treatment of SARS-CoV-2 based on previously records of their therapeutic efficacy in SARS-COV and MERS-COV [38,39].Recent computational studies showed that SARS-CoV-2 Mpro structure had four residues for lopinavir binding (M49, M165, P168, and Q189), nine residues for ritonavir binding (L27, H41, M49, F140, N142, G143, H164, M165, and E166) and seven residues for remdesivir binding (Thr25, Thr26, His41, Phe140, Asn142, Cys145, His163) [40,41]. In contrast, our study identifies eight residues (HIS41, Phe140, GLY143, CYS145, HIS163, HIS164, GLU166 and Q189) as active functional sites showed potential binding activities. In addition to that, some nsSNPs related to these residues predicted to be highly dameging to the protein, which would make these residues suitable sites for drug targeting. Moreover, a newly in vitro experimental study shows powerful activity of remdesivir and lopinavir dagainst SARS-CoV-2 [42,43]. Successful case reports describing the partially effective of remdesivir for the early confirmed patients of SARS-CoV-2 infection [44-46]. Despite these findings, a clinical randomized controlled trial performed on adult hospitalized patients with severe SARS-CoV-2, no benefits of lopinavir/ritonavir or Remdesivir treatment were observed [47-49]. This variability in effectiveness of HIV protease inhibitors might be relies on that HIV protease is belongs the aspartic protease enzymatic class, whereas SARS-CoV-2 Mpro belongs to the cysteine protease enzymatic class, do not contain a C2-symmetric pocket, which is the target of HIV protease inhibitors. Accordingly, on the basis of this evidences, the current antiviral drugs need to be modified in order to be effective againsSARS-CoV-2 infection.
5. Conclusions
- The computational approach can significantly reduce the financial cost and time for drug development against the SARS-COV-2 Mpro. In this study, five residues (HIS41, GLY143, CYS145, HIS163 and GLU166) predicted as active functional sites showed potential binding activities. Thirty four high-risk damaging nsSNPs of SARS-COV-2 Mpro were related to these residues resulting in stability and structural changes of proteins and changes in the interactions between domains, thus affecting the function of CoV-2 Mpro. These characteristics provide them the promising to be target sites for the fresh generation inhibitors to work with and overcome the current drugs ineffectiveness.Combining methods such as site-directed mutagenesis assay and nuclear magnetic resonance potentially help scientists to better understanding the effect of this predicted SNPs on SARS-COV-2 Mpro structural stability and functional offering basis to develop specific drugs for inhibiting this virus.
ACKNOWLEDGEMENTS
- The authors acknowledge Dr. Ahmed Abdelbagi Hammad for language editing and proofreading of the manuscript.