-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Virology and Molecular Biology
p-ISSN: 2163-2219 e-ISSN: 2163-2227
2015; 4(1): 4-11
doi:10.5923/j.ijvmb.20150401.02
Molecular and Phylogenetic Analysis of Hepatitis C Virus and Its Implication in Lagos State
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLFaeji Charles Oluwafemi 1, Omilabu Sunday 2
1College of Medicine, Afe babalola University, Ado Ekiti, Nigeria
2College of Medicine, University of Lagos
Correspondence to: Faeji Charles Oluwafemi , College of Medicine, Afe babalola University, Ado Ekiti, Nigeria.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Hepatitis C infection is a progressing and devastating live threatening health care problem in developing and developed countries worldwide caused by hepatitis C virus. The virus belongs to the family Flaviviridae and genus hepacivirus. Hepatitis C virus continues to be a disease burden in Nigeria and the world at large, and man is known to be its natural host. However, Information on genotype of HCV in Nigeria is limited, hence the need for more work. This study will help to determine the prevalent genotype as this will have influence on management of patients and expected therapeutic response because response to treatment varies according to genotype and treatment may prevent progression of infection to hepato-cellular carcinoma. This research was conducted with 40 sero-positive blood samples consisting of 23 females and 17 males. The samples were analyzed by Reverse transcriptase- Polymerase Chain Reaction (RT- PCR) and two samples were positive for HCV RNA. This extracted viral genome was sequenced and a phylogenetic tree was constructed from its analysis. This phylogenetic analysis showed that the strain did not cluster with known strains from the zone hence suggestive of a possible new strain or unidentified strain or strain yet to be reported in this zone, hence the recommendation for expanded and further research. The RT-PCR test confirmed the presence of hepatitis C virus in Lagos and that sero-positive samples may not definitely be positive for RT- PCR test.
Keywords: Molecular, Phylogenetic Analysis, Hepatitis C virus, Lagos State, Implications
Cite this paper: Faeji Charles Oluwafemi , Omilabu Sunday , Molecular and Phylogenetic Analysis of Hepatitis C Virus and Its Implication in Lagos State, International Journal of Virology and Molecular Biology, Vol. 4 No. 1, 2015, pp. 4-11. doi: 10.5923/j.ijvmb.20150401.02.
Article Outline
1. Introduction
- Hepatitis C infection is an infectious disease affecting primarily the liver and it is caused by hepatitis C virus (HCV) [1]. The virus is an RNA virus that belongs to the family Flaviviridae and genus hepacivirus [1]. Hepatitis C Virus is a single strand enveloped RNA virus, with eleven genotypes [2].Hepatitis is an inflammatory condition of the liver while viral hepatitis is a conventional term used to denote hepatitis caused by the hepatotrophic viruses (hepatitis A-G). The disease presents a serious public health problem in developing countries like Nigeria and the world at large, affecting over 170 million people [3]. HCV has been discovered to play a primary role in post transfusion hepatitis, establishment of long standing persistent infections and association with chronic cirrhosis and hepato-cellular carcinoma [4, 5]. HCV accounts for 20% of cases of acute hepatitis, 70% of cases of chronic hepatitis, 40% of cases of end-stage cirrhosis, 60% of cases of hepatocellular carcinoma (HCC) and 30% of cases of liver transplants [1]. Other causes of chronic Liver diseases are viral hepatitis (hepatitis B and D, cytomegalovirus, Epstein Barr virus), toxoplasmosis, schistosomiasis, inherited and metabolic disorders, drugs and toxins. Endemicity in the developing parts of the world is high with majority of people being sero-positive, with most becoming infected at birth or in early childhood [5].As a result of mutations, the genomes of HCV strains show extensive variability. However, some regions of the genome are more variable than others, and classification of HCV genotypes is based on differences in the variable regions of the genome. HCV is divided into eleven phylogenetically distinct groups 1-11 designated as clades (groups of genotypes that share a common ancestor) [6]. Within the clades, a number of subtypes (individual genotypes) have been defined [7]. Genotype 1b has been found in Nigeria and 4 have also been reported [8, 9]. Genotype 1 and 3 is most prevalent in the USA [10]. HCV found in the Coast region of Africa is known to be genotype 1 [8]. Genotypes 2 and 4 are majorly found in sub-Saharan Africa (Gabon, Cameroon, Uganda, Egypt) [11].All known types of HCV have the potential to cause serious liver disease [10]. There are various hepatitis virus causing liver disease, they include Hepatitis A, B, C, D, E viruses. There is strong evidence demonstrating the association of chronic HCV infection to cirrhosis and hepato-cellular carcinoma (HCC). HCV is a mounting global health challenge, causing a significant proportion of chronic liver disease around the world. Hepatitis C can be transmitted through Sexual contact, Inoculation with contaminated blood or blood products, Transplantation of organs from infected donors, Parenterally from infected mothers, Intravenous drug use [6] and symptoms include lethargy, anorexia, nausea, abdominal discomfort, alcohol intolerance, though sometimes shows no symptoms in the early stage and in a progressed and advanced stage it includes increased lethargy, fluid retention, bruising, prolonged bleeding, peripheral stigmata of CLD, gynaecomastia, Ascites/oedema, splenomegaly, distended abdominal Veins, bruising, hepatic encephalopathy, jaundice [6].Diagnosis of hepatitis C infection could be achieved by Serology using ELISA method, detection of viral protein by western blot and also by detection of viral particle by RT-PCR.As blind treatment is not encouraged, treatment should be based on genotype because response to treatment varies according to genotype [12]. There is difficulty in treatment of HCV genotype 1 as against its other genotypes for example and this ineffective treatment may allow the progression of infection to HCC [12]. Drugs used include Ribavirin, Pegylated interferon, Alpha Interferon and duration of treatment ranges between24weeks – 48weeks (genotype 1b) and no vaccine is available for yet for HCV [6].Improved understanding of the rate of nucleotide sequence mutation in HCV has allowed the approximate time of divergence of major genotypes to be designated as 1-11 [6] and the origin and spread of the present epidemic of hepatitis C to be better defined [13]. Improved methods of genotype over the last few years have enabled recognition of the importance of genotype in the progression of HCV-related liver disease and response to anti-viral therapy to be studied [1]. Present data strongly indicates that HCV genotype is an important determinant of response to treatment and a result of the advances in methodology and recent results of large clinical trials of combination therapy, knowledge of HCV genotype is now central to the clinician in the management of patients with chronic hepatitis [14]. Genotype 1b has mostly been implicated for hepatocellular carcinoma (HCC), however it has a considerable fair prognosis with treatment [14]. It is estimated that up to 50 percent of the people with genotype 1b will have a sustained response, or successful treatment, with pegylated interferon and ribavirin [12]. Patients with genotypes 2 and 3 are almost three times more likely than patients with genotype 1b to respond to therapy with alpha interferon or the combination of alpha interferon and ribavirin [12]. Furthermore, when using combination therapy, the recommended treatment length depends on the genotype. For patients with genotypes 2 and 3, a 24-week course of combination treatment is adequate, whereas for patients with genotype 1b, a 48-week course is recommended [12]. For these reasons, testing for the genotype of hepatitis C is useful [11, 12] and helps in determining the prevalent genotype.Hepatitis C virus continues to be a disease burden in Nigeria and the world at large, and man is known to be its natural host.This study will help to determine the prevalent genotype as this will have influence on management of patients and expected therapeutic response because response to treatment varies according to genotype and treatment may prevent progression of infection to hepato-cellular carcinoma [12].However, Information on genotype of HCV in Nigeria is limited [8], hence the need for more work.Therefore this study was aimed at determining the genotype of Hepatitis C virus in Lagos by detecting the presence of Hepatitis C viral RNA in HCV sero-positive samples, sequencing the detected viral genome and further constructing a phylogenetic tree to compare the strain with reference strains using molecular method
2. Materials and Methods
- ● STUDY DESIGN: A study on hepatitis C virus in Lagos● STUDY SITE: Virology laboratory, Central research Laboratory, Idi Araba.● STUDY POPULATION/SIZE: 40 HCV sero-positive blood samples consisting of 23 females and 17 males (ethical approval was obtained for this study).● MATERIALS: Disinfectant (Hypochlorite, spirit), Hand gloves (powdered and latex powdered free), Foil papers, Eppendorf tubes, Centrifuge, Vortex machine, Incubators, Weighing balance, Conical flask, Thermocycler (9800 Gold plated Applied Biosystem (ABI) PCR machine) , Refrigerator -60℃, -80℃, -8℃ (Thermocool), Water distiller, Bio-safety cabinet (applied biosystem), biosafety cabinet, Electrophoresis tanks, Trans-illuminator/imager, Autoclave.
2.1. Laboratory Analysis
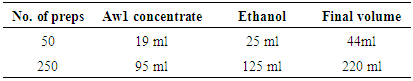
- SAMPLE PROCESSINGPreparation of reagentsAddition of carrier RNA to Buffer AVLi. 310 μl of Buffer AVE was added to the tube containing 310 μg lyophilized carrier RNA to obtain a solution of 1 μg/μl. ii. The carrier RNA was thoroughly dissolved and divided into conveniently sized aliquots, and stored at –20°C. The aliquots carrier RNA was not freeze–thawed for less than 3 times.iii. The buffer AVL was checked for precipitate, and incubated at 80°C when necessary until the precipitate was dissolved. The volume of Buffer AVL–carrier RNA mix needed per batch of samples was based on the number of samples to be simultaneously processed. For numbers of samples, volumes were calculated using the following sample calculation:n x 0.56 ml = y mlyml x 10 μl/ml = z μln = number of samples to be processed simultaneouslyy = calculated volume of Buffer AVLz = volume of carrier RNA–Buffer AVE to add to Buffer AVLBuffer AW1● Buffer AW1 was supplied as a concentrate. Before using for the first time, appropriate amount of ethanol (96–100%) as indicated on the bottle, in the Table below.
|
|
2.2. Protocol (Qiagen) for Extraction of viral RNA
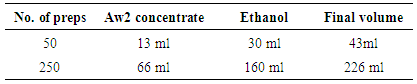
- Viral RNA was extracted using QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany).Procedure:● 560 μl of prepared Buffer AVL containing carrier RNA was aliquot into a 1.5 ml micro centrifuge tube.● 140 μl of serum was added to the Buffer AVL–carrier RNA in the micro centrifuge tube. It was mixed by pulse-vortex machine for 15 s.● Incubation was done at room temperature (15–25°C) for 10 min.● The tube was briefly centrifuged to remove drops from the inside of the lid.● 560 μl of ethanol (96–100%) was added to the sample, and mixed by pulse-vortex machine for 15 s. After mixing, the tube was briefly centrifuged to remove drops from inside the lid.● 630 μl of the solution from the above step was carefully applied to the QIAamp Mini column (in a 2 ml collection tube) without wetting the rim. The cap was closed and centrifuged at 6000 x g (8000 rpm) for 1 min. The QIAamp Mini column was placed into a clean 2 ml collection tube. The tube containing the filtrate was discarded.● The QIAamp Mini column was carefully opened, and above step was repeated.● The QIAamp Mini column was carefully opened, and 500 μl of Buffer AW1 was added. The cap was closed, and centrifuged at 6000 x g (8000 rpm) for 1 min. The QIAamp Mini column was placed in a clean 2 ml collection tube (provided). The tube containing the filtrate was discarded.● The QIAamp Mini column was carefully opened and 500 μl of Buffer AW2 was added. The cap was closed and centrifuged at full speed (20,000 x g; 14,000 rpm) for 3 min. ● The QIAamp Mini column was placed in a new 2 ml collection tube, and the tube containing the filtrate was discarded. It was then centrifuged at full speed for 1 min.● The QIAamp Mini column was placed in a clean 1.5 ml micro centrifuge tube. The old collection tube containing the filtrate was discarded. The QIAamp Mini column was carefully opened and 60 μl of Buffer AVE was added. The cap was closed, and incubation was done at room temperature for 1 min. It was then centrifuged at 6000 x g (8000 rpm) for 1 min.● Viral RNA was stored at –20°C or –80°C.
|
|
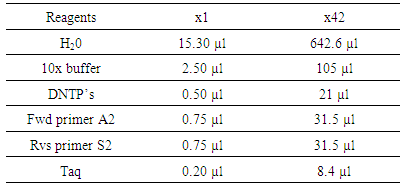
 Resulting amplicons were stored at -8ºc refrigerator and further run on gel electrophoresis.
Resulting amplicons were stored at -8ºc refrigerator and further run on gel electrophoresis.2.3. Gel Electrophoresis
- Preparation of gel electrophoresisi. A 1.5g of agarose gel was weighed with a weighing balance and poured in a conical flask and mixed with 100ml of Tri Acetate EDTA (TAE) buffer.ii. The conical flask was heated in a microwave until the powder was completely dissolved and the gel allowed to cool to a temperature tolerated by the cheek.iii. 1.5 µl of sybr safe solution was then added and rocked gently for to solution to mix properly. iv. The mixture was poured into a gel casting glass containing two combs placed at distance apart and allowed to solidify.v. On solidification, the combs were removed gently and the cast was placed in the electrophoresis tank and tae buffer was poured to cover the cast.vi. 1 µl of loading buffer was mixed with 5 µl of amplicon, in a loading tray and dispensed into the various wells in the gel.vii.2 µl of the ladder was placed in the 8th well, while the negative and positive controls were put in the last 2 wells. viii.The gel was then run at 120volts for 20mins and the result was viewed using a trans-illuminator.ix. The expected base pair of 200bp was then compared with that of the ladder or markerto check for positives.
2.4. Pcr Clean up
- PCR clean up was done using NucleoSpin Gel and PCR Clean-up kit (Manufactured by MACHEREY-NAGEL GmbH & Co. KG, Düren · Germany).Protocol:i. A clean scalpel was used to excise the DNA fragment from the agarose gel. All excess agarose was removed. The weight of the gel slice was determined and transferred to a clean tube. ii. Sample was incubated using heating block (BioBlock Scientific, Thermolyne Corporation, Iowa, USA) for 5–10 min at 50°C. The sample was mixed (Vortex mixer- Vision Scientific Co. Ltd, Korea) briefly every 2–3 min until the gel was completely dissolved.iii. NucleoSpin® Gel and PCR Clean-up Column was placed into a Collection Tube (2 ml) and 700 μl of sample was loaded.iv. It was centrifuged for 30s at 11,000rpm. The flow-through was discarded and the column was placed back into the collection tube.v. 700 μl Buffer NT3 was added to the NucleoSpin® Gel and PCR Clean-up Column. It was again centrifuged for 30s at 11,000rpm. The flow-through was discarded and the column was placed back into the collection tube.vi. It was centrifuged for 1 min at 11,000rpm to remove Buffer NT3 completely. It was ensured that the spin column did not come in contact with the flow-through while removing it from the centrifuge and the collection tube vii. The NucleoSpin Gel and PCR Clean-up Column was placed into a new 1.5 ml micro-centrifuge tube.viii. 30 μl Buffer NE was added and incubated at room temperature (18–25°C) for 1 min and centrifuged for 1 min at 11,000rpm.
2.5. Sequencing and Genotyping
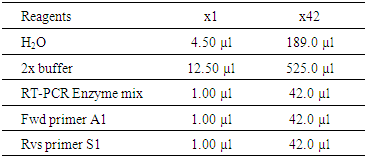
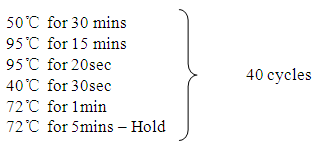
- i. Raw DNA was analyzed and sequencedii. It was compared and aligned with reference sequence using the BASIC LOCAL ALIGNMENT SEARCH TOOL (BLAST) by National Center for Biotechnology information (NCBI) targeting the 5’ un-translated region (UTR).iii. Phylogeny and molecular evolutionary analyses were conducted using maximum likelihood, evolutionary distance and maximum parsimony method of Molecular Evolutionary Genetics Analyses (MEGA) [15].iv. A phylogenetic tree was constructed with bootstrapping of 1000 replicate method.
3. Result
- Of the 40sero-positive samples consisting of 17 males and 23 females were analyzed, two samples were positive for HCV RNA and according to the phylogenetic tree, the genotype does not cluster with the reference strains from the region.
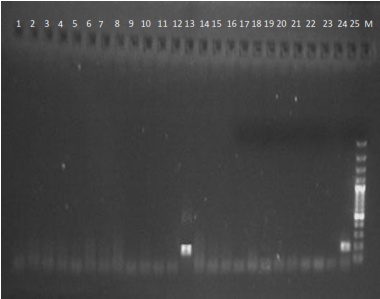 | Figure 1. Detection of HCV RNA in serum samples as shown by Gel electrophoresis, showing (samples 1 – 25) a positive result with band size of 200bps |
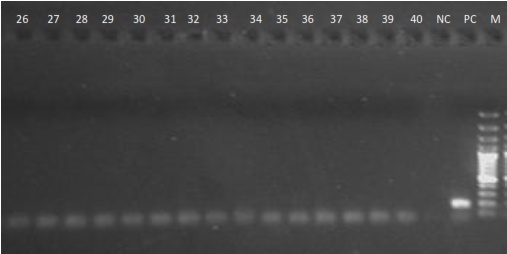 | Figure 2. Detection of HCV RNA in serum samples as shown by Gel electrophoresis, showing samples 26 - 40 |
 | Figure 3. Molecular phylogenetic analysis using maximum likelihood, evolutionary distance and maximum parsimony method showing the clusters of Lagos strains, Ibadan strains and the reference strains |
4. Discussion
- The essence of further research on hepatitis C virus cannot be overemphasized because HCV is a major causative agent of hepatitis which accounts for 20% of cases of hepatitis infection, 70% of cases of chronic hepatitis, 40% of cases of end stage cirrhosis and 60% of cases of hepato-cellular carcinoma., especially in developed countries. Screening of blood by serology is the common method for detecting HCV but PCR has proven to be most sensitive. This study has shown that the detection of HCV particle in two samples of the antibody sero-positive samples tested was attainable by qualitative RT-PCR test and the genotype found was related to reference strains of HCV. Of 40 samples tested with PCR, two were positive, that is not all sero-positive samples can be positive for PCR which is in conformity with the research of [16] who studied 350 ELISA positive samples and 6 were positive for PCR and also carried out PCR assay on 338 RIBA positive samples but none positive for PCR.It was also reported by [17], on evaluation of 79 samples that were EIA positive and for which HCV RNA was not detected by PCR, that is PCR negative, which he attributed to low HCV nucleic acid concentration or HCV titer in the samples to be detected and also infection clearance and that individuals may just be carriers and virus is not actively replicating in them. The samples positive for PCR, that is viral RNA was detected implies that the individual has an active or ongoing infection and there is active viremia [18].Active viremia is an indication of the replication of the virus and sometimes an evidence of chronic HCV infection [18]. PCR is the most sensitive assay but antibody screening may prove useful in measuring past infections or present, irrespective of the actual infectivity of the individual. However, not all anti HCV positive are PCR positive because the level of viremia may be too low for detection by the current PCR assay, poor storage of samples and treatment of patients may also have deleterious effect on PCR result [19]. The immune response to HCV infection is important in whether the infection would overwhelm the individual or the infection would be cleared. The negativity for PCR might also be due to dormancy or no replication of the virus in the host or most possibly, clearance of HCV infection as a result of host immune system [20]. The detection of HCV RNA by RT-PCR permits direct detection of the presence of the virus and also detection during sero-negative window period, immediately after infection, therefore implying that the detection of HCV RNA may be more reliable than serology especially in patients who do not mount enough antibody response due to impaired immune system [18, 20]. Despite the expertise, proper sample handling, good laboratory conduct and effective protocol used to carry out this study, two of 40 sero-positive samples were positive for PCR which means that the patients are actively infected and has potential infectivity. This does not eliminate the possibilities of potential infectivity from other PCR negative/serology positive individuals [20]. Antibodies may have been produced due to past infection and antibody produced will still be detected by serology but may not be detected by PCR because PCR detects viral genome [20]. The sequencing result as shown in the phylogenetic tree (fig 3) shows that the sequence of the positive strains are not related to any established reference strain. It is also seen that the sequences of Ibadan samples clustered with the samples from Lagos but the two samples from Lagos did not cluster with the reference strains.This could mean that the sequences of the samples fall in a genotype or strain that has not yet been reported or established before in the region or a possible new strain or not in the reference comparison. Therefore, further work needs to be done. However limitations encountered during this study is inclined towards the fact that RT-PCR is experience and expertise required, also expensive and limited facilities are available, therefore it is mostly routinely done for samples positive by serology. Due to the prevalence of this infection and disposition of individuals towards its diagnostic tests, limited number of samples could be collected over a period of months for the research.
5. Conclusions and Recommendations
- The RT-PCR test confirmed the presence of hepatitis C virus in Lagos and that sero-positive samples may not definitely be positive for RT- PCR test. Both PCR and serology could be used to detect hepatitis C infection but PCR is more sensitive and should be carried out for individuals.This research is suggestive of a possible new strain or unidentified new strain yet to be reported in this region, hence the need for expanded and further work which will lead to the establishment of a prevalent strain in the area as this will significantly assist in the management of HCV infections in the state and prevent progression to liver cancer. It is recommended that HCV infections should be taken more seriously because it could be severe in its chronic form, and should be included as a serious blood borne pathogen which should be tested for before any blood transfusion in health care facilities in the state. The health system too should be empowered to support all HCV infected persons medically and socially, as well as supporting research in vaccine development. People positive for serology tests should be tested for HCV RNA and sequencing be done to determine the genotype for effective management because response to treatment varies according to genotype. Further and expanded research should to be done to ascertain the prevalent genotype and underline the impact of movement of high risk population on changing molecular epidemiology.
ACKNOWLEDGEMENTS
- I acknowledge the effort and supervision of Professor Omilabu S.A over this research work and appreciate the financial and moral support of my sponsors, Mr. and Mrs. A.O Faeji.I gratefully acknowledge the contributions of the following to this study; Dr. Salu O. B, Mr. James Ayotunde, Mrs. Remi, Joseph Shaibu, Damola Oresanya, Moyinoluwa Okedusi and Banji Oso.