-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Tumor Therapy
p-ISSN: 2163-2189 e-ISSN: 2163-2197
2020; 9(1): 1-4
doi:10.5923/j.ijtt.20200901.01

Adoptive Cellular Immunotherapy for Solid Tumors
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLNader Hazboun
Biology Department, Bethlehem University, Bethlehem, Palestine
Correspondence to: Nader Hazboun, Biology Department, Bethlehem University, Bethlehem, Palestine.
| Email: |  |
Copyright © 2020 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Adoptive cellular therapy (ACT) has shown promise for the treatment of cancer specifically the use of chimeric antigen receptor (CAR) T cell therapy for leukemias and lymphomas. But the translation of this success to solid tumors remained elusive. Solid tumors are characterized by an immunosuppressive microenvironment that renders immune effector cells inactive and exhausted. This review is focused on describing the various strategies of adoptive cellular therapy for solid tumors including tumor infiltrating lymphocytes (TILs), T cell receptor (TCR) transduced T-cells, CAR-T cells, natural killer (NK) cells, and gamma-delta (γδ) T cells. The role of combination therapy with checkpoint inhibitors to reverse tumor resistance is also discussed.
Keywords: Immunotherapy, T cells, Checkpoint inhibitors
Cite this paper: Nader Hazboun, Adoptive Cellular Immunotherapy for Solid Tumors, International Journal of Tumor Therapy, Vol. 9 No. 1, 2020, pp. 1-4. doi: 10.5923/j.ijtt.20200901.01.
Article Outline
1. Introduction
- Adoptive cellular therapy using interleukin (IL)-2 expanded tumor infiltrating lymphocytes has shown promising results in the treatment of advanced cancer and, in particular, for a subset of patients refractory to standard therapy [1,2]. These findings, while not applicable to all tumors, led to the development of novel methods to introduce anti-tumor TCRs and CARs into autologous lymphocytes for therapeutic use (Fig. 1) [2]. TCRs can be designed to target self-antigens that are minimally expressed in normal body tissues and are highly overexpressed in tumors such as Wilms tumor antigen 1 and MART-1 that are upregulated in melanoma [3]. CAR T-cell therapy has shown exciting results in CD19 expressing hematologic malignancies, but how to translate this to solid tumors will require overcoming several therapeutic barriers, chief among them is the immunosuppressive tumor microenvironment which incorporates many factors that can limit an effective immune response even when tumor antigens are successfully identified [3,4]. Natural killer (NK) cells are very effective at mediating cytotoxicity against tumor cells and unlike T-cells, kill their targets in a non-major histocompatibility complex (MHC)-antigen specific manner and without the need for prior sensitization, and are being assessed in clinical trials but their efficacy has been so far modest at best [1]. There is clear evidence that γδ-T cells impact the progression of human tumors, either as natural immune surveillance or as therapeutic agents [5]. In this review, the different strategies of adoptive cellular therapy for solid tumors are summarized together with combination therapy with checkpoint inhibitors.
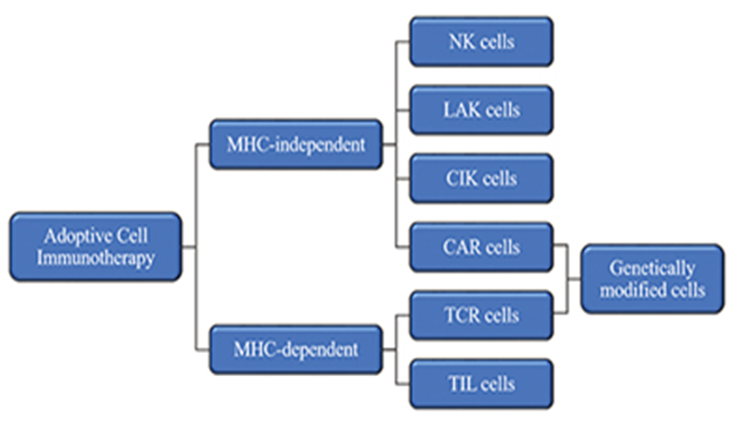 | Figure 1. Effector cells for adoptive cellular immunotherapy. Abbreviations: MHC, major histocompatibility complex; NK, natural killer; LAK, lymphokine-activated killer; CIK, cytokine-induced killer; CAR, chimeric-antigen receptors; TCR, T cell receptors; TIL, tumor-infiltrating lymphocytes [16] |
2. Strategies of Adoptive Cellular Therapy
2.1. Tumor Infiltrating Lymphocytes
- Tumors are commonly infiltrated by T cells directed against tumor-associated antigens and neoantigens. These T cells are extracted from tumor biopsies, undergo IL-2 mediated or anti-CD3, anti-CD28, and allogeneic feeder cells mediated ex-vivo expansion, and then reinfused into patients after receiving lymphodepleting preconditioning regimen including non-myeloablative cyclophosphamide and fludarabine chemotherapy or the addition of total body irradiation (TBI) [6]. The clinical trials conducted on patients with metastatic melanoma showed that transfer of expanded TILs and recombinant IL-2 preceded by a cyclophosphamide and TBI regimen doubled the complete response rate (CR) to 24% [7,8]. Recently, preliminary data showing objective and complete responses were reported for different solid tumors including cholangiocarcinoma, cervical cancer, gastrointestinal, gynecological, head and neck, breast and lung cancers [7].
2.2. TCR-transduced Lymphocytes
- As an alternative to the process of isolating TILs is to genetically modify T cells isolated from the blood to express transgenic TCRs able to recognize tumor antigens such as the cancer-testis antigen NY-ESO-1 which induced objective clinical responses in patients with synovial cell sarcoma, melanoma, or myeloma, without severe toxicity, and this is likely to be FDA-approved [9]. Other TCRs that could be transduced into T cells target lineage restricted antigens such as MART, gp100, or Melan A, but one limitation of targeting these antigens is their potential to also target normal cells, as evidenced by vitiligo observed in patients treated with MART-specific T cells [10]. Identification of TCRs with reactivity against immunogenic neoepitopes that can be genetically introduced into autologous T cells is being actively explored for development of personalized therapies. However, one of the major limitations of transgenic TCRs is that they are dependent on external co-stimulation and presentation of neoepitopes via MHC-I complex which is often downregulated in cancer cells [9].
2.3. CAR-transduced Lymphocytes
- Chimeric antigen receptors (CARs) are genetically engineered receptors consisting of an antigen-binding domain that is derived from a single-chain variable fragment (scfv) of a monoclonal antibody, a flexible spacer/hinge region, a transmembrane domain, a CD3-ζ or Fc-γ intracellular signaling domain and one or two costimulatory domains such as CD28,4-1BB, or OX40 [11]. There have been an increasing number of CAR T-cell trials for solid tumors because of their unprecedented efficacy for hematological malignancies. These CARs target various tumor-associated antigens or tumor-specific oncogenic mutations such as truncated MUC-1, EGFRvIII, mesothelin, HER-2, IL13Rα, glypican-3, PSMA, and ErbB2 [12]. Early studies attempting to translate CAR-modified T cells to solid malignancies have been hampered by expression of the target on normal tissues. For example, a patient with ERBB2 (HER2/Neu)-positive metastatic colorectal carcinoma, who was tested with ERBB2-redirected CAR T cells, developed respiratory distress within 15 minutes of treatment followed by cardiac arrest and death. Postmortem analysis suggested localization of CAR T cells to the lung with activation triggered by low levels of ERBB2 on lung epithelial cells [4]. Thus far, however, clinical trials with CAR T-cells have in addition to target selection, T cell proliferation and persistence, also specific hurdles to overcome, such as trafficking of the T cells into the tumor and the ability to overcome an immunosuppressive microenvironment [13].
2.4. Natural Killer Cells
- Natural killer (NK) cells are a population of innate lymphoid cells (ILCs) that can induce the death of allogeneic and autologous cells undergoing malignant transformation or microbial infection. Many efforts have been made to exploit NK cells in clinical practice, and more than 200 clinical trials have been carried out with the aim of potentiating the effector capacities of these cells in vivo [14]. NK cells express an array of activating receptors such as NKG2A and inhibitory receptors such as NKG2D on their cell surface, therefore strategies to block inhibitory receptors, enhance activating receptors, or redirect their specificity through CAR transduction can be used to enhance killing of target cells [15]. In patients with advanced renal cancer, several phase I trials of ex vivo expanded allogeneic NK-92 cells, an immortalized NK cell line, have already reported effective anti-tumor activity [6]. CAR-engineered NK cells are being explored to target B-cell malignancies, breast and ovarian cancer, colon cancer, prostate cancer and sarcoma [1]. In an alternative NK-based therapeutic approach, immune stimulatory cytokines such as IL-2 and IL-15 are administered to patients to induce activation and expansion of the autologous NK cell population [14].
2.5. Cytokine-induced Killer (CIK) Cells
- CIK cells are a heterogeneous CD8+ T cell population induced by the addition of anti-CD3, interferon-γ, and IL-2 ex vivo. A phase II trial to determine whether the combination of radiofrequency ablation and cytokine-induced killer cell infusion can prolong survival in patients with ovarian cancer is underway (NCT02487693) [16].
2.6. Gamma-delta (γδ) T Cells
- γδ T cells share many qualities with their αβ counterparts, such as cytotoxic effector functions and pro-inflammatory cytokine production, but one major difference is their major histocompatibility complex molecule (MHC) antigen recognition independence. The mechanisms by which γδ T cells kill cancer cells are similar to those of conventional cytotoxic T cells and involve release of perforin and granzyme B, and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and FAS ligand (FASL) expression inducing apoptosis in the target cells [5]. A recent study showed that MART-1 and gp-100 reactive γδ1+ TCRs bind to human leukocyte antigen A2 (HLA-A2) identifying an MHC-restricted γδ TCR which opens up the possibility for using these cells in adoptive cell therapies [17].
3. Obstacles to Adoptive Cellular Therapy
3.1. Solid Tumor Microenvironment
- The presence of an aberrant tumor vasculature with adhesion molecule down-regulation, FASL-upregulation, and inhibitory receptors in addition to an unfavorable chemokine profile prevent homing of effector cells to the tumor bed [16,18]. Within the solid tumor microenvironment, tumor cells can upregulate a variety of inhibitory receptors like programmed cell death ligand 1 (PD-L1), and secrete molecules including IL-10, transforming growth factor β (TGFβ), and prostaglandin E2 (PGE2), that can directly block T cell function and/or attract and activate immunosuppressive cells including T-regulatory cells (Tregs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and tumor associated neutrophils (TANs) [18]. TAMs in turn can express PD-L1 and PD-L2 which bind the T cell inhibitory receptor programmed cell death protein 1 (PD-1). TAMs and Tregs also secrete immunosuppressive cytokines such as IL-10 and TGFβ [19]. In addition, solid tumors frequently demonstrate metabolic aberrations that can impact T cell function and survival including depletion of arginine and tryptophan due to production of arginase and indoleamine 2,3 dioxygenase 1 (IDO-1) respectively by TAMs and MDSCs and also have hypoxia leading to an extracellular matrix acidification due to insufficient vascular supply [4].
3.2. Cytokine Release Syndrome
- Cytokine release syndrome (CRS) is a potentially life-threatening toxicity that is mostly reported after ACT infusion in hematological malignancies but has been reported for melanoma [3]. It is caused by massive release of inflammatory cytokines such as IL-6, and IFN-γ by the infused T cells or the endogenous macrophages that participate in the anti-tumor immune response [20]. Tocilizumab, an anti-IL-6 receptor antagonist, is used to control CRS without affecting the anti-tumor activity of the T cells [21]. Systemic corticosteroids are also effective against CRS but they may inhibit ACT efficacy and therefore should be used mainly in cases of severe neurological symptoms or after tocilizumab failure [3].
3.3. On-Target Off-Tumor Toxicity
- On-target off-tumor toxicity occurs when a targeted tumor antigen (e.g. HER2) is also expressed on other tissues at a level that can still be recognized by the engineered T cell [22]. If these antigens are expressed in vital organs such as the heart, liver, and kidney, they will cause fatal damage [16]. Exciting new strategies are being developed to engineer MHC-independent T cells that recognize multi-antigen signatures or antigen densities, which have the potential to dramatically improve tumor recognition specificity [22].
4. Combination Therapy with Checkpoint Inhibitors
- T cells express various receptors on their surface that can either stimulate T cell activity or inhibit T cell responses and can be used by tumors to evade the immune system. PD-L1, for example is overexpressed in a number of cancers, leading to impaired T cell antitumor responses via PD-L1-PD-1 interactions. Blocking this interaction systemically using anti-PD-L1 and/or anti-PD-1 monoclonal antibodies (mAbs) has shown remarkable success in the clinic [23]. CAR T cells from biopsies of glioma patients demonstrated increased expression of several immunosuppressive markers such as Foxp3 and PD-1 opening up the possibility for combination therapy with checkpoint inhibitors [24]. In a clinical trial involving a sub-cohort of 11 patients with malignant pleural mesothelioma who received cyclophosphamide preconditioning followed by a single dose of mesothelin-directed CAR T cells and subsequent anti-PD-1 agent (at least 3 doses) with a minimum of 3 months of follow-up, 8 patients showed response (response rate, 72%), including complete metabolic responses in 2 patients. An ongoing phase I clinical trial at the University of Pennsylvania is exploring the combination of EGFRvIII-directed CAR T-cells with the humanized anti-PD-1 antibody pembrolizumab in patients with glioblastoma [25]. An alternative approach to merging adoptive therapy with checkpoint blockade is to modify T cells to express cytotoxic T-lymphocyte associated antigen-4 (CTLA-4) or PD-1/CD28 chimeric molecules where upon ligand binding, transforms the inhibitory CTLA-4 or PD-1 signal into a positive stimulatory signal, and this innovative approach has resulted in maximized T-cell anti-tumor function in preclinical lymphoma and melanoma syngeneic mouse models [26].
5. Conclusions & Future Directions
- It is now well established that T cells are potent “living drugs” in cancer therapy and have been extensively studied for various types of cancer. Although TCR and CAR modified T cells have shown great potential in treating some cancer types, the major obstacle to further development is the solid tumor immunosuppressive microenvironment [23]. This could be tackled by using combinatorial strategies of ACT with immune checkpoint inhibitors which showed great potential in preclinical models and in clinical trials. Other combinatorial approaches could involve ACT with inhibitors of MDSCs, TAMs, T-regs, IDO, and TGFβ. CAR-T cells can induce substantial toxic effects and the manufacture of the cells is complex. These adverse effects could be mitigated by using “off-the-shelf” CAR-transduced NK cells.