-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Tumor Therapy
p-ISSN: 2163-2189 e-ISSN: 2163-2197
2017; 6(1): 1-4
doi:10.5923/j.ijtt.20170601.01

Cancer Immunotherapy with CAR-T Cells for Hematological Malignancies
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLNader Hazboun
Biology Department, Bethlehem University, Bethlehem, Palestine
Correspondence to: Nader Hazboun, Biology Department, Bethlehem University, Bethlehem, Palestine.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Cancer treatment for hematological malignancies traditionally included systemic chemotherapy and localized radiotherapy for central nervous system (CNS) disease. These therapies in addition to their adverse effects’ profile do not induce complete responses in all patients. The development of chimeric antigen receptor (CAR) technology enables the targeting of tumors with high affinity and specificity leading to unprecedented responses among refractory/relapsed patients with various blood malignancies. This mini-review summarizes the role of CAR-T cells for the treatment of blood cancer highlighting potential areas of improvements.
Keywords: Immunotherapy, Monoclonal antibody, Chimeric antigen receptor T-cells
Cite this paper: Nader Hazboun, Cancer Immunotherapy with CAR-T Cells for Hematological Malignancies, International Journal of Tumor Therapy, Vol. 6 No. 1, 2017, pp. 1-4. doi: 10.5923/j.ijtt.20170601.01.
Article Outline
1. Introduction
- Cancer treatment for hematological malignancies traditionally included systemic chemotherapy and localized radiotherapy for central nervous system (CNS) disease. These therapies in addition to their adverse effects’ profile do not induce complete responses in all patients. A fourth modality for cancer treatment involves immunotherapy. It involves either passive or active means of stimulating the immune response against cancer. Results of treatment with monoclonal antibodies and nonmyeloablative alloHSCT demonstrate that immunotherapy can be effective in patients with B-cell malignancies, but much room for improvement remains. The development of immunotherapies with greater efficacy than monoclonal antibodies and less toxicity than alloHSCT would be a major advance in treatment of hematological malignancies [1]. Despite skeptical views on its role against cancer, the recent successes of checkpoint inhibitors and chimeric antigen receptor T-cells has fueled unprecedented enthusiasm in the field. The development of chimeric antigen receptor (CAR) technology enables the targeting of tumors with exquisite affinity and specificity leading to complete responses among refractory/relapsed patients with various blood malignancies [1]. This mini-review summarizes the role of CAR-T cells for the treatment of blood cancer highlighting trials with the best results and potential areas of improvement.
2. CAR Structure and Design
- The basic structure of a first generation chimeric antigen receptor (CAR) consists of an antigen-recognition domain named single chain fragment variable (scFv) derived most commonly from a monoclonal antibody, and an intracellular domain derived either from a CD3ζ chain or an FcRγ chain (Fig.1A) [2]. As a master T-cell co-stimulator, CD28 engagement amplifies TCR signaling when the T-cell receptor (TCR) is also engaged by cognate peptide-major histocompatibility (MHC) complex [3]. Telomere length and telomerase activity/activation are modulated by CD28 signaling, which implies that reduced CD28 expression is associated with shortening of telomeres and decreased telomerase activity [4]. Second generation CARs involves the addition of a co-stimulatory signal from CD28 or 4-1BB molecules (Fig. 1B) while third generation CARs have two co-stimulatory signals in their intracellular domain (Fig. 1B) [2]. T-cells expressing anti-CD19 CARs with CD3ζ signaling domains and co-stimulatory domains such as CD28 or 4-1BB had enhanced antigen-specific cytokine production, proliferation and survival compared with CARs with only CD3ζ signaling resulting in more effective eradication of human leukemia cells [1]. The co-stimulatory signaling moieties used in third-generation CARs include CD28, 4-1BB, or OX-40 (CD134). In pre-clinical studies, they have demonstrated superior anti-tumor efficacy compared with second-generation CAR T-cells [5]. The CAR T-cell through its scFv binds to a cell surface antigen of a given tumor in a major-histocompatibility complex (MHC)-independent manner. Therefore, the CAR T-cells are not susceptible to common mechanisms of tumor escape, including downregulation of MHC molecules and aberrant antigen processing, and are less sensitive to micro-environmental inhibitory factors [2, 5]. The CAR construct is introduced into T-cells by using a retroviral or a lentiviral vector. This enables long-term expression of the CAR in the T-cell, in contrast to the transposon system of transduction which results in transient expression of the CAR [2].
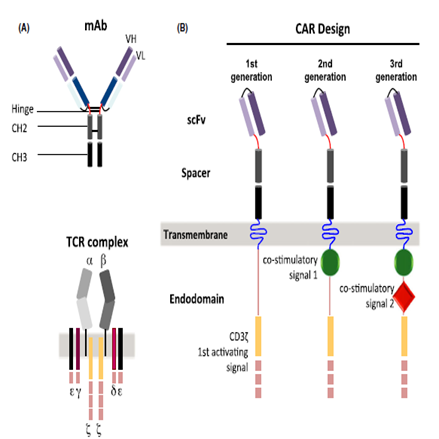 | Figure 1. Chimeric antigen receptor (CAR) design. (A) Structural components of monoclonal antibody (mAb) and T cell receptor(TCR) used in the construction of a CAR are shown. (B) Structures of CARs of different generations [2] |
3. Clinical Uses of CAR T-cells
- Clinical trials of CAR T-cell therapy for the treatment of cancer have mostly been conducted in patients with CD19-positive hematological malignancies, such as B-ALL, CLL, follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL), and mantle-cell lymphoma (MCL) [5]. Ongoing trials are also being conducted in multiple myeloma patients. The use of CAR T-cells in each of these diseases is presented in the following sections with emphasis on trials with the best results.
3.1. Acute Lymphoblastic Leukemia
- The best clinical results have been obtained with CD19-targeted CAR T-cells against acute lymphoblastic leukemia. Clinical trials were performed in different centers across the US [5]. The highest response rates were obtained at Memorial Sloan Kettering Cancer Center (MSKCC), in which the study involved 32 adults with relapsed/refractory B-ALL using a second generation CAR (CD28, CD3ζ) (NCT01044069) [6]. The complete response rate was 91% with B-cell aplasia and cytokine release syndrome as major toxicities [6].
3.2. Chronic Lymphocytic Leukemia
- The clinical trial at the National Cancer Institute (NCI) involved 4 patients with CLL who received pre-conditioning lymphodepleting chemotherapy consisting of fludarabine and cyclophosphamide and 1-5 x 106/Kg of CAR T-cells. The overall response rate (ORR) was 100% (3 patients with complete response (CR), and one patient with partial response (PR). The duration of response (DoR) ranged from 4-22 months [7].
3.3. Non-Hodgkin’s Lymphoma
- Researchers at MSKCC reported on 10 evaluable patients with relapsed, aggressive histology NHL [8]. The protocol investigated administering CAR T cells in the setting of high-dose chemotherapy (BEAM regimen; carmustine, etoposide, cytarabine, melphalan) followed by autologous stem cell rescue. Six out of 10 evaluable patients achieved a complete response, with four of them remaining free from disease progression after 13-21 months of follow-up [8].
3.4. Hodgkin’s Lymphoma
- CD30 was chosen as a target for CAR therapy in HL because it is only expressed on Reed-Sternberg cells, the malignant cells of this disease and on a subset of activated CD45RO+ T-cells [9]. A phase I trial at Baylor college of medicine is actively recruiting patients with Hodgkin and Non-Hodgkin lymphoma to study the side effects and the best dose of CD-30 expressing CAR T-cells (NCT01316146) [10].
3.5. Multiple Myeloma
- B-cell maturation antigen (BCMA) is a cell-surface receptor that belongs to the tumor necrosis superfamily and its expression is restricted to plasma cells. For this reason, BCMA-based therapies might result in eradication of plasma cells, both cancerous and nonmalignant, an ‘ontarget, off-tumor’ toxicity that can be effectively managed clinically with IVIg. The expression pattern of BCMA and results of preclinical studies have indicated that BCMA is an attractive target for CAR therapy of MM [5]. Currently, a phase I trial at the National Cancer Institute (NCI) is actively recruiting patients with previously treated multiple myeloma to determine the safety and feasibility of anti-BCMA CAR T cells (NCT02215967) [11].
4. Toxicity & Immunogenicity
- The major toxicity seen in the CAR19 T cells trials has been attributed to severe cytokine storm seen in conjunction with rapid T-cell proliferation. It is caused by the toxic release of pro-inflammatory cytokines such as IL-6 [12]. Tocilizumab, an anti-IL-6R antibody has been used in the clinic to dampen the effects of cytokine release syndrome (CRS) in many patients [5]. The potential immunogenicity of the transduced CAR must be considered. Some of the scFvs that are incorporated in the CARs used in clinical trials are of murine origin and can elicit an anti-IgG or even IgE response. Because of this many groups are now using human or humanized scFvs in their CAR constructs [12].
5. CAR Modifications & Combinations
- CARs have been genetically modified to express IL-12. This eliminated the need for preconditioning lymphodepletion, and rendered the CARs resistant to inhibition by regulatory T-cells in a syngeneic model of CD19+ malignancies [13]. On target, off tumor activation of engineered T-cells has been associated with dose-limiting toxicities and death in some cases. Therefore, deletion of CAR T-cells in case of toxicity in animal models has been achieved using an inducible human caspase9 (icasp9), which can be induced by intravenous administration of FK506 binding protein (FK506BP) [14]. Advanced CAR designs may also provide another level of safety. For example, the inclusion of a second inhibitory CAR specific for antigens expressed on normal but not tumor cells, will allow safer targeting by the activating CAR of the antigens expressed by both tumor and normal cells [2]. In an effort to promote CAR trafficking, GD2-specific CAR T-cells have been engineered to co-express the chemokine receptor CCR2b in order to enhance tumor infiltration and anti-tumor efficacy [15]. Approaches aimed at neutralizing specific immunoregulatory mechanisms or the addition of immune checkpoint inhibitors such as anti-CTLA-4 and anti-PD1 antibodies to CAR T-cell therapy approaches may overcome immune suppression and enhance CAR T-cell mediated tumor regression [16].
6. Conclusions & Future Perspectives
- The current results of CAR T-cell clinical trials in hematologic malignancies clearly demonstrate the efficacy of this approach in improving the outcome of patients with relapsed/refractory leukemias or lymphomas. Many patients have already obtained complete remissions that have been long-lasting in patients with advanced-stage B-cell malignancies using anti-CD19 CAR T cells [1]. The rapidly evolving technology in the genetic engineering of CAR T cells enables the modification of the toxicity profile, the trafficking, and the efficacy of this powerful immunotherapeutic approach so that it will become an important standard treatment for many hematologic malignancies.