-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Tumor Therapy
p-ISSN: 2163-2189 e-ISSN: 2163-2197
2015; 4(1): 1-4
doi:10.5923/j.ijtt.20150401.01
A Rare Case of Multicentric Castleman Disease Secondary to Alemtuzumab Therapy
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLPrajwal C. Boddu M. D.1, Abdalla Hassan MD2, Ann Mauer M. D.3, Deepti A. Singh M. D., Elliot S. Weisenberg M. D.
1Internal Medicine, Advocate Illinois Masonic Medical center, Chicago, USA
2Internal Medicine, Advocate Illinois Masonic Medical center
3Oncology-Hematology, Creticos cancer center, Advocate Illinois Masonic Medical Center4Pathology, Advocate Illinois Masonic Medical Center
Correspondence to: Prajwal C. Boddu M. D., Internal Medicine, Advocate Illinois Masonic Medical center, Chicago, USA.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Multicentric Castleman disease (MCD) is a rare group of lymphoproliferative proliferative disorders. Human herpes Virus 8 has been shown to play a central role in the pathogenesis of the disease, and is present in most cases of HIV+ MCD patients and few cases of HIV-MCD patients. The etiology of HIV-, HHV8-MCD remains unknown. A 49 year old African American male presented to our hospital with 2 week history of fevers and night sweats. He participated in an experimental Alemtuzumab drug trial, four years ago for Multiple Sclerosis. At the time of admission, all hematological indices were normal. On physical examination, patient had signs of supraclavicular lymphadenopathy and generalizededema. Over the hospital course, he developed stable leukocytosis, normocytic anemia and thrombocytopenia. Labs were notable for an elevated ESR, CRP and LDH. Computed Tomogram showed diffuse lymphadenopathy and hepatomegaly. Axillary lymph node histopathology was consistent with MCD. The patient's serum tested negative for HIV and HHV8. He was started on a 4 week regimen of Rituximab and Etoposide. He showed good clinical response to the first cycle of the regimen and is currently in clinical remission. Our case is only the second case report of MCD reported as a delayed serious adverse event after being on Alemtuzumab therapy. Our case illustrates the fact that Alemtuzumab therapy can be associated with serious delayed drug effects, hence requiring close vigilance. Our patient responded to Rituximab and Etoposide combination and this regimen should be considered as a treatment option in such patients.
Keywords: Castleman Disease, Alemtuzumab
Cite this paper: Prajwal C. Boddu M. D., Abdalla Hassan MD, Ann Mauer M. D., Deepti A. Singh M. D., Elliot S. Weisenberg M. D., A Rare Case of Multicentric Castleman Disease Secondary to Alemtuzumab Therapy, International Journal of Tumor Therapy, Vol. 4 No. 1, 2015, pp. 1-4. doi: 10.5923/j.ijtt.20150401.01.
1. Introduction
- Castleman disease (CD) is a heterogeneous group of benign lymphoproliferative disorders with a wide clinical spectrum, ranging from asymptomatic lymph node enlargement to B-symptoms with extensive diffuse lymphadenopathy and hepatosplenomegaly. Castleman disease has traditionally been classified into localized (LCD) and metacentric (MCD) model of disease based on clinical presentation. Increasing recognition of Castleman disease as a pathomorphological syndrome has allowed for a histopathological classification into atleast three variants based on morphology of the follicle centers and plasmacytosis: -1) Hyaline vascular variant (HVV) 2) Plasma cell variant (PCV) 3) Plasmablastic variant [1]. HVV constitutes upto 90% of unicentric disease with the plasma cell variant constituting almost all the cases of multicentric disease [1, 2]. Hyaline vascular variant is characterized morphologically by atretic, hyalinized follicular centers with lymphocytosis and follows a more indolent course. The PCV, characterized by hyperplastic germinal centers and plasmacytosis, commonly presents with constitutional B symptoms and hepatosplenomegaly, irrespective of its centricity [2].Kaposi Sarcoma Herpes Virus/HHV-8 virus has been recognized to play a pivotal role in the pathogenesis of the disease, and is present in almost all cases of HIV+ MCD patients and a few cases of HIV-MCD patients. Most of HIV-MCD patients are HHV8 negative, and the etiology of MCD in this group of patients remains obscure. This report describes a case of HHV8-MCD, 4 years after initiation of Alemtuzumab, an exploratory immunotherapy being considered for the treatment for Multiple Sclerosis.
2. Case Report
- A 49 year old African American male presented to our hospital with 2 week history of fevers, sweats and persistent abdominal pain. 10 years prior, he suffered an attack of Multiple Sclerosis (MS), and has been on multiple treatments ever since. He participated in an experimental Alemtuzumab trial therapy, four years prior, with abatement of neurological symptoms since the completion of the regimen, three years ago. He has not been on any immunosuppressant since then. At the time of admission, all hematological indices were normal. Alkaline phosphatase was high at 156IU/L with mild hypoalbuminemia (2.6g/dL) and increased globulin concentrations (4.6g/dL). Two days into admission, he underwent right Hemicolectomy after developing Pneumatosis Intestinal is of unclear etiology. Post-operative course was complicated by severe sepsis, acute tubular necrosis requiring blood pressure support, broad spectrum antibiotics and temporary hemodialysis. On physical examination, patient had a toxic appearance and there were signs of bilateral supraclavicular lymphadenopathy and generalized edema. Over the hospital course, he developed stable leukocytosis without bands (12000/mm3), normocytic normochromic anemia (Hb 7.4g/dL) and thrombocytopenia (98x109/L). Labs were notable for an elevated ESR (>120mm/hr), CRP (>20mg/L) and LDH(425U/L). Computed Tomogram demonstrated diffuse (mesenteric, axillary, mediastinal) lymphadenopathy, bilateral pleural effusions and mild hepatomegaly (Fig A, B). Thoracentesis fluid was transudative and without malignant cells. Urinary protein excretion was 1.5g/24 hr and Bence Jones protein was not detected. Urine immunofixation studies showed a polyclonal increase in κ and λ chains, with no M protein. The following investigations were either negative, non-reactive or within normal range: autoantibodies, Cryoglobulin, complement, c-ANCA, p-ANCA, Hepatitis panel, RPR, blood, urine and pleural fluid cultures. Because of a strong suspicion for lymphoproliferative disorder, an axillary lymph node biopsy was undertaken. Histology of the lymph node revealed atretic follicles with Plasmacytosis, prominent paracortical vascularity and sinus Histiocytosis with an overall appearance consistent with Multicentric Castleman disease (Fig C, D). Antibodies to various antigens were negative for monoclonal Bcells and T cells with abnormal antigenic expression. Immunohistochemistry for K and L chains showed polyclonal activation of Plasma cells and immunohistochemistry for Spirochetes and HHV8 was negative. Based on clinical and pathological suspicion for Castleman disease, the following labs were ordered and revealed normal IL-6(<5pg/ml), normal IgG4(23mg/dL) and high VEGF(351pg/ml). The patient's serum tested negative for HIV and HHV8. He was diagnosed with HIV negative, HHV8 negative Multicentric Castleman disease of Plasma cell variant. After an informed consent was obtained, he was started on a 4 week regimen of Rituximab and Etoposide. He showed good clinical response to the first cycle of the regimen and is currently in clinical remission. He is to get a PET scan, one month from the time of this writing.
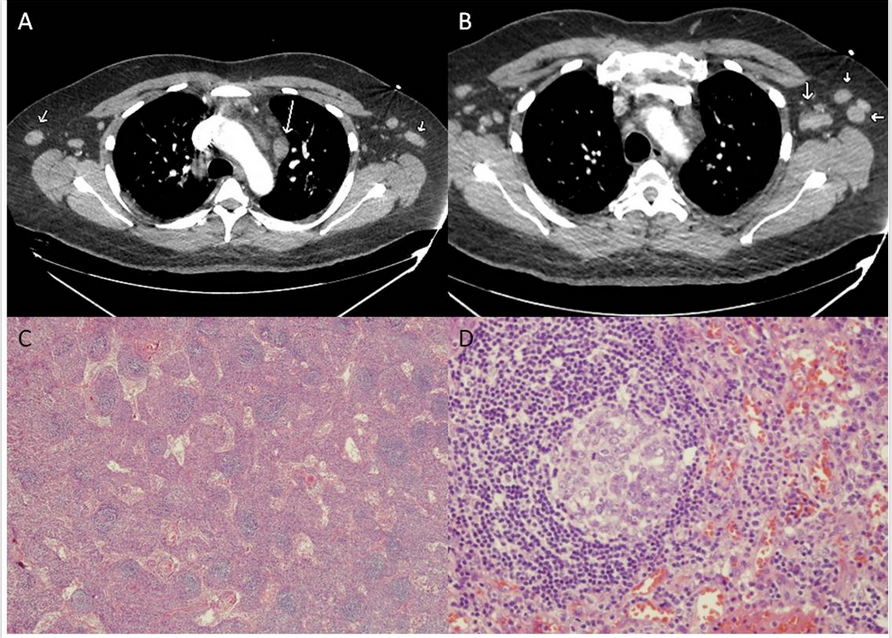 | Figure. (A) Axial CTA image of the chest initially performed for shortness of breath showing several enlarged bilateral axillary lymph nodes (small arrows), enlarged prevascular lymph node (long arrow). (B) Few prominent, axillary lymph nodes (small arrow). (C) Axillary node biopsy-original magnification X 40, atretic and few hyperplastic lymphoid follicles. (D) Original magnification X 400, mildly hyperplastic follicle with prominent mantle zone |
3. Discussion
- MulticentricCastleman disease is an uncommon benign lymphoproliferative disorder characterizedby lymph node hyperplasia with Plasmacytosis and vascular proliferation, fever, anemia, hypergammaglobulinemia, thrombocytosis/ thrombocytopenia, and an increase in plasma levels of acute phase reactants. Advances in the understanding of the pathophysiology of the disease have shown HHV8 to be central to the causation of MCD, with HHV8 reported in almost all the cases of HIV+MCD and up to 40% of the cases of HIV- MCD [3]. The pivotal etiological role of HHV8 in the pathogenesis of MCD is assuming practical importance in terms of prognosis and management, and has led to restructuring of the classification of MCD based on HHV status into HHV8+ and HHV8- subclasses [4]. HHV8 infected MCD patients tend to have a more clinically aggressive course than HHV8- MCD [5, 6]. The etiology of HHV8- MCD remains obscure, with various uncharacterized processes being proposed based on varying lines of evidence, which include systemic inflammatory, paraneoplastic and non-HHV8 viral driven mechanisms [4]. Other markers central to the pathogenesis of MCD are IL-6 and VEGF [7, 8]. A simplified unifying pathophysiology model is that HHV-8 infected plasma cells encode viral cytokine (vIL-6) and produce VEGF, inducing intranodal vascular proliferation and endothelial production of human IL-6 [7, 8, 9, 10]. Detection of vIL-6 expression in lymph nodes has prognostic implications with vIL-6 positive Castlemandisease patients having a more fatal clinical course [6]. IL-6 hypercytokinemiais responsible for the intranodal Plasmacytosis and for the systemic signs and symptoms seen in MCD [11].MCD is associated with KaposiSarcoma (KS), HIV, POEMS syndrome and should be considered in patients at high risk for MCD (KS, HIV, Primary effusion lymphoma) [2, 12]. PCV MCD has fairly non-specific histopathological features of hyperplastic germinal centers with Plasmacytosis, and it is important to exclude secondary causes of reactive lymphadenopthy, such as drug reactions, lymphomas, autoimmune conditions and systemic inflammatory processes. A diagnosis of MCD is entertained based on clinical symptomatology and characteristic histopathology after an exhaustive exclusion of other reactive processes. IL-6 is characteristically high in MCD, and may be used to follow treatment response since symptoms and biochemical abnormalities wax and wane depending on fluctuating IL-6 levels [7]. However, IL-6 has a short half-life of less than 6 hours and this may account for the normal IL6 levels detected in our patient's blood, which might have been drawn during a waning period of the disease course [13]. Our case has some of the specific findings for an emerging disease concept in Japan, the TAFRO syndrome [thrombocytopenia, anasarca, fever, Reticulin fibrosis and organomegaly], including Thrombocytopenia, pleural effusions and anasarca, which can’t be explained solely by increased IL-6 production and may well be due to other cytokine storms [14]. However, etiology and pathogenesis of TAFRO syndrome is unknown and it is still not clear if it is a distinct clinical and pathological entity or a part in a certain process of MCD [9]. MCD also shares many clinical and pathological features with IgG4 lymphadenopathy; however the normal IgG4, high CRP and low platelet count help reliably differentiate MCD from IgG4 lymphadenopathy in our case [15].Heterogeneity of presentation and rarity of incidence accounts for the paucity of clinical trials and treatment is based largely upon experiences in observational studies and case series reports. Case studies have found Rituximab, a monoclonal anti CD20 antibody, to be effective with durable remissions in HIV- MCD and may be considered as first line option in MCD [16, 17]. Progressive disease requires addition of chemotherapy, allowing for an immunochemotherapeutic approach in single agent and multi-agent chemotherapy combinations depending upon performance status. Single combination chemotherapy regimens found to effective in MCD patients include oral Etoposide [18] chemotherapy, and may be given to poor performance status patients harboring progressive disease. The more toxic multi-agent CVD (Cyclophosphamide, Vincristine, Doxorubicin) and CHOP (Cyclophosphamide, Adriamycin, Oncovin, Prednisone) chemotherapy regimens follow clinical experiences with treatment for non-Hodgkin’s Lymphoma and are reserved for suitableMCD patients with good performance status [3, 17]. Our patient was started on combination of Rituximab and Etoposide therapy on a 4 week regimen. He responded well to the regimen and is currently in clinical remission. He is to get a PET scan a month from the time of this writing. Alemtuzumab is an anti-CD52 humanized monoclonal antibody undergoing experimental trials for the treatment of MS. Clinical trials have shown Alemtuzumab to significantly more effective in suppressing relapses than the traditional Interferon-Beta therapy [19]. However, significant issues with Alemtuzumab therapy include malignancies and delayed autoimmune reactions months to years after treatment [19, 20]. In an extended phase 2 trial involving a SM3 pilot study, one patient developed MCD 31 months after the initiation of Alemtuzumab therapy, and has been in remission to date after treatment with R-CHOP [20]. Our patient developed MCD 47 months after initiation of therapy.
4. Conclusions
- Multicentric Castleman disease is rare lymphoproliferative disorder which can occur in the absence of HIV infection. This is only the second case report of MCD reported as a delayed serious adverse event after being on Alemtuzumab therapy. Alemtuzumab use requires careful monitoring for early detection of potential delayed adverse events. Our patient responded to Rituximab and Etoposide regimen and this should be considered as a treatment option in such patients. Siltuximab, an investigational monoclonal anti IL-6 antibody blocking the function of IL-6, has also been shown to be effective for the treatment of HIV negative Multicentric Castleman disease in a phase 2 trial study and was recently approved by the FDA for the same [21].
ACKNOWLEDGEMENTS
- The authors would like to thank Dr. Michael Gioia, for his valuable help with Radiology images.