-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Tumor Therapy
p-ISSN: 2163-2189 e-ISSN: 2163-2197
2014; 3(1): 1-9
doi:10.5923/j.ijtt.20140301.01
In Vitro Anticancer Activity of Monosubstituted Chalcone Derivatives
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLVisakh Prabhakar 1, Ranganathan Balasubramanian 2, Priyanka Sathe 3, C. Murali Krishna 3, Aarti Juvekar 4
1Department of Pharmacy, Birla Institute of Technology and Sciences, Pilani Campus, Rajasthan-333031, India
2Department of Pharmaceutical Chemistry, Amrita School of Pharmacy, Amrita Vishwa Vidyapeetham University, Health Sciences Campus, Kochi, Kerala-682041, India
3Chilakapati Laboratory, Cancer Research Institute (CRI), Advanced centre for Treatment, Research and Education in Cancer (ACTREC), Tata Memorial Centre (TMC), Kharghar, Navi Mumbai-410210, India
4Anti-Cancer Drug Screening Facility (ACDSF), Advanced centre for Treatment, Research and Education in Cancer (ACTREC), Tata Memorial Centre (TMC), Kharghar, Navi Mumbai-410210, India
Correspondence to: C. Murali Krishna , Chilakapati Laboratory, Cancer Research Institute (CRI), Advanced centre for Treatment, Research and Education in Cancer (ACTREC), Tata Memorial Centre (TMC), Kharghar, Navi Mumbai-410210, India.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Cancer is a continuously adapting dynamic condition for which investigation of novel treatment strategies constitutes an integral component in its management. As part of the continuing efforts from the global community directed at eradication of cancer, we were interested in evaluating the cytotoxic potential of a series of chalcone derivatives. The study also contributes towards a preliminary understanding of the structure-activity relationship of synthetic chalcone derivatives. We screened various A-ring monosubstituted chalcone derivatives against five selected human cancer cell lines using the sulforhodamine B assay. Our in vitro observations indicated the previously established characteristic biphasic response of the chalcones. Moreover, it was found that the different substitutions at the A-ring were found to suppress the cytotoxic activity, which was evident from higher growth suppression by the unsubstituted chalcone compared to the substituted derivatives. The compound 4c exhibited similar activity to that of the unsubstituted derivative, indicating that the methyl group is not detrimental to activity. It was observed that the tested compounds were most effective on the A2780 ovarian cancer cell line exhibiting GI50 value ranging from 20-66 µM. Moreover, the analogs proved to be least cytotoxic in the HepG2 cell line with GI50 values that ranged from 39-100 µM. This study may act as a preliminary step towards understanding substituent effects on the cytotoxic activity profiles of chalcone derivatives emphasising on A-ring substituted moieties.
Keywords: Chalcones, Cytotoxicity, Cancer Cell Lines, Sulforhodamine B assay
Cite this paper: Visakh Prabhakar , Ranganathan Balasubramanian , Priyanka Sathe , C. Murali Krishna , Aarti Juvekar , In Vitro Anticancer Activity of Monosubstituted Chalcone Derivatives, International Journal of Tumor Therapy, Vol. 3 No. 1, 2014, pp. 1-9. doi: 10.5923/j.ijtt.20140301.01.
Article Outline
1. Introduction
- A large number of anticancer drugs currently in clinical practice are either plant-derived or synthetic derivatives of plant metabolites, including clinically significant classes of cytotoxic compounds like taxols, vinca alkaloids, and other similar molecules. Chalcones, which constitute a privileged group of compounds, are biosynthetic precursors of naturally occurring flavonoids and isoflavonoids found in a variety of plant species. They possess a 1,3-diaryl-prop-2-ene-1-one scaffold in which the two aromatic rings (labelled A and B) are linked by an α,β-unsaturated carbonyl system (Figure 1). Quercetin, xanthohumol, isoxanthohumol, genistein, naringenin and chalconaringenin are some of the well known pharmacologically active members of this class of natural products. Specific examples of antiproliferative chalcones found in nature include isoliquiritigenin, isobavachalcone, xanthoangelol and licochalcone A[1]. Extended conjugation and a high degree of electrophilicity associated with the moiety play an important role in the activity of this class of compounds[2-4]. The complete delocalization of π electrons on both the benzene rings makes these compounds more susceptible in undergoing electron transfer reactions that may be attributed to their good antioxidant activity. It has been indicated that the s-trans conformation is more active in demonstrating cytotoxic activity than the s-cis conformation [3]. The extremely simple skeleton of chalcone compared to the complex structures of most other anticancer drug candidates makes this scaffold very attractive to chemists for alteration and structure-activity relationship (SAR) modifications. The diverse pharmacological activity of this class of compounds has drawn the attention of medicinal chemists and pharmacologists alike for a long time. Chalcone derivatives have shown anticancer, antimalarial, antiprotozoal, anti-inflammatory, antibacterial, antifilarial, antifungal, antimicrobial, anticonvulsant and antioxidant activity[5-14]. The anticancer activity of chalcone derivatives has been a topic of interest due to the association of specific functional groups and substituents to a particular activity or lack thereof against a specific cell line. For instance, the radical quenching activity has been firmly associated with the phenolic hydroxy groups[15-17].One of the most widely studied antiproliferative mechanisms of action of chalcone derivatives is microtubule destabilization that prevents the polymerization of tubulin protein to form microtubules that are essential for mitotic activity[18, 19]. SAR studies have confirmed that chalcone derivatives with electron-donating hydroxy or methoxy groups act in this fashion. Chalcones bind to the colchicine binding site on the β subunit of tubulin, thus triggering depolymerization[20, 21]. Chalcone derivatives have been found to be significantly active in growth inhibition and induction of apoptosis in prostate cancer cell lines. Besides, they have also been studied for their chemopreventive effects by sensitizing the cells to TRAIL TNF[(Tumor Necrosis Factor)-related apoptosis-inducing ligand]-mediated apoptosis[21, 22]. Studies have indicated that the cytotoxicity of chalcones is related linearly to their ability to form phenoxy radicals by oxidation, as well as to their hydrophilicity[23]. In particular, the cytotoxic mechanisms of the hydroxy derivatives were attributed to the mitochondrial uncoupling and subsequent interference with the cellular respiration[24]. Apart from the apoptosis phenomenon, many other possible mechanisms like inhibition of tubulin assembly, inhibition of angiogenesis, anti-estrogenic activity, and reversal of multi drug resistance (MDR) have been attributed to the anticancer activity of this chosen class of compounds[15, 16]. The present study was carried out on a mini selection of monosubstituted chalcones in order to get a preliminary understanding of the role of substituents, with respect to their electronics as well as regiochemistry, on the antitumor activity of the chalcone skeleton.
2. Materials and Methods
2.1. Reagents and Chemicals
- NaHCO3 (Sigma), 10 mM minimal essential medium (MEM) non-essential amino-acid solution (Invitrogen), 100 mM sodium pyruvate (Hyclone), FBS (PAA Laboratories), MEM (Eagle) supplemented with 2 mM L-glutamine and Earle’s balanced salt solution (MEM/EBSS; Hyclone), 10 mg/mL bovine insulin in 25 mM HEPES, pH 8.2 (Sigma), 2.5% (wt/vol) trypsin solution (Invitrogen), 0.5% (wt/vol) phenol red solution (Sigma), 0.48 mM versene-EDTA, 0.4% (wt/vol) trypan blue in 0.81% (wt/vol) NaCl and 0.61% (wt/vol) KH2PO4 (Sigma), dimethyl sulfoxide (DMSO; Sigma), 10% (wt/vol) trichloroacetic acid (TCA), 10 mM unbuffered Tris base solution.
2.2. Design and Synthesis of Chalcone Derivatives
- Five derivatives were carefully designed based on medicinal chemistry principles. Since the objective of the study was to understand substituent effects on the activity, the unsubstituted chalcone (4a) formed the base compound. In order to realize the stated goal, the plan was to append substituents of different chemical nature at each of the three available regioisomeric positions on the A-ring. The task of choosing the substituents involved identifying functional groups that withdraw as well as release electrons both by inductive and mesomeric effects. The target derivatives included 2’-methylchalcone (4c), 3’-nitrochalcone (4f), 4’-methoxychalcone (4h), and 4’-bromochalcone (4j). While the alkyl and halo groups in 4c and 4j release and withdraw electrons inductively (+I and –I respectively), the nitro and alkoxy groups in 4f and 4h do so via resonance (-M and +M respectively). Regiochemistry was a crucial design consideration as these four derivatives comprising of an ortho (represented by carbon atoms 2’ and 6’ in Figure 1) congener, a meta (represented by carbon atoms 3’ and 5’ in Figure 1)-substituted chalcone and two para (represented by carbon atom 4’ in Figure 1) analogs account for all the available positions in the A-ring. These four monosubstituted chalcones together with the parent moiety 4a complete the set of target compounds in this study. Since this work is hoped to serve as a starting point for systematic SAR investigation, five additional chalcone derivatives [2’-hydroxychalcone (4b), 2’-bromochalcone (4d), 2’-aminochalcone (4e), 3’-bromochalcone (4g), and 4’-phenylchalcone (4i)] were prepared following the same design criteria. Based on the preliminary findings reported herein, all these ten analogs would be subjected to further in depth studies in order to facilitate greater understanding of structure function correlation.
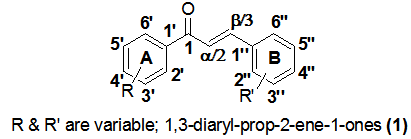 | Figure 1. Basic chalcone skeleton with substituted rings A and B |
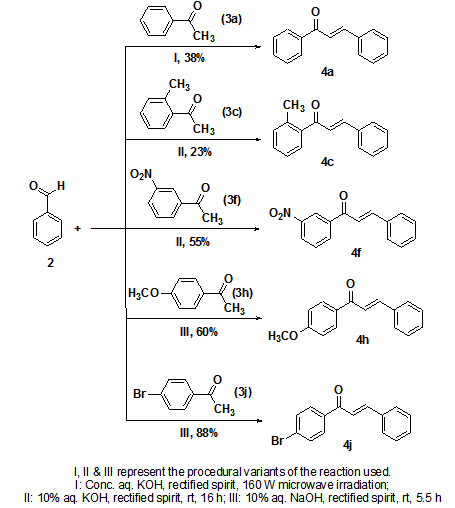 | Scheme 1. Synthesis of target chalcone derivatives |
2.3. Cell Culture and In Vitro Cytotoxicity Assay
- Cytotoxicity of the five compounds was estimated in different human cell lines using the sulforhodamine B (SRB) protocol[25]. Five human cell lines - U373MG (human glioblastoma cell line), MCF-7 (human breast cancer cell line), HepG2 (human pancreatic cancer cell line), K562 (human myelogenous leukemia cell line) and A2780 (human ovarian cancer cell line) were selected for this study. Cells were inoculated into 96-well microtiter plates (90 µL/well) at appropriate plating densities depending on the doubling time of individual cell lines. The microtiter plates were incubated at 37 °C in a carbon dioxide (CO2) incubator at 5% CO2, 95% air and 100% relative humidity for 24 h prior to addition of experimental drugs. The cells were then incubated for 48 h with the test compounds. The experiment was terminated by adding 30% chilled TCA to the wells. Cells were stained using 0.4% SRB in 1% acetic acid. The SRB dye bound to the fixed cells was eluted using 10 mM unbuffered Tris solution. The optical density was measured using ELISA micro plate reader (Tecan-Sunrise) at a wavelength of 540 nm with 690 nm reference wavelength. Percent growth was calculated on a plate-by-plate basis for test wells relative to control wells and was expressed as the ratio of average absorbance of the test well to the average absorbance of the control wells multiplied by 100. Using the six absorbance measurements[time zero (Tz), control growth (C), and test growth in the presence of drug at the four concentration levels (Ti)], the percentage growth was calculated at each of the drug concentration levels. Percentage growth inhibition was computed using the formulae:
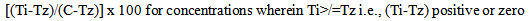 | (1) |
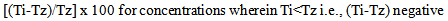 | (2) |
2.4. Statistical Analysis
- All values were expressed as mean ± standard error of mean (SEM). All statistical analysis was performed using Graph Pad (Version-3) Prism software. Data for cytotoxicity studies were analyzed using one way Analysis of Variance (ANOVA) followed by Dunnetts test. A confidence interval of 95% was selected and P<0.05 was considered statistically significant compared to the control group.
3. Results and Discussion
3.1. Cytotoxic Parameters
- The results of the cytotoxicity assay exhibit an interesting trend across all the cell lines. Almost all the chalcone derivatives were found to be active only at 10-4M concentration. Moreover there is no linear response between the drug concentration and cell death. While the 10-4M concentration exhibited significant activity, the 10-5M concentration did not exhibit an activity that was even comparable to the former. The same trend was found in the other higher dilutions also. This interesting phenomenon is in conformation with a previously reported study that attributes this trend to the characteristic biphasic response of chalcone derivatives[26]. In the case of the U373MG cell lines, 4h was found to be the least active while 4a exhibited highest cytotoxicity. It is evident that the compounds 4c and 4j exhibited higher cytotoxicity than the reference compound Adriamycin (Figure 2). In MCF-7 breast cancer cell line, derivatives 4a and 4c exhibited the highest activity; however, consistent with the previous neuronal cell line, the cytotoxic activity showed a significant increase at 10-4 M concentration (Figure 3).
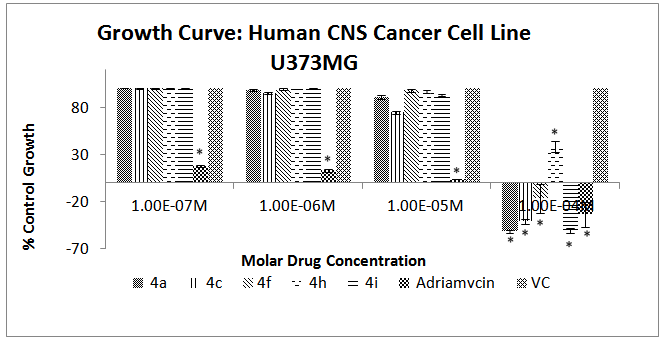 | Figure 2. Effect of different chalcone derivatives on the percentage growth inhibition of the U373MG cell line[Values are Mean ± SEM of 3 replicates each. *P<0.05, VC = Vehicle Control (DMSO)] |
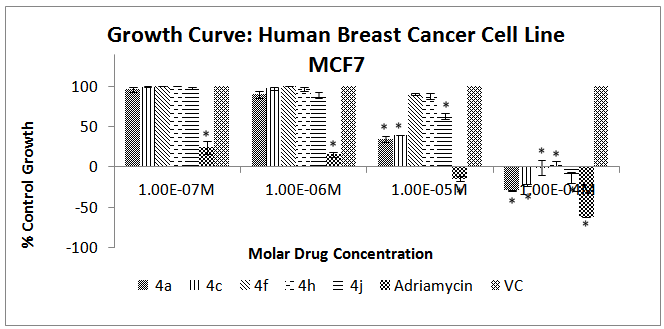 | Figure 3. Effect of different chalcone derivatives on the percentage growth inhibition of the MCF-7 cell line[Values are Mean ± SEM of 3 replicates each. *P<0.05, VC = Vehicle Control (DMSO)] |
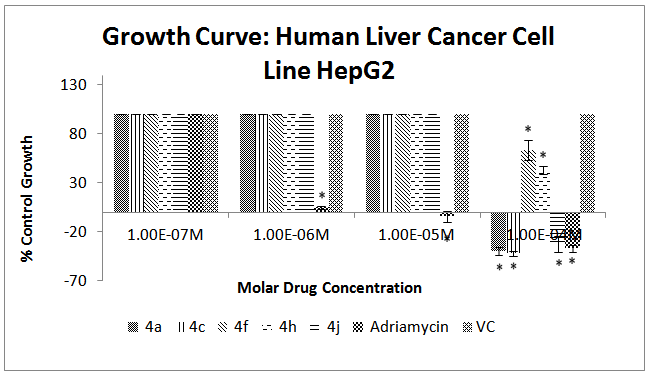 | Figure 4. Effect of different chalcone derivatives on the percentage growth inhibition of the HepG2 cell line[Values are Mean ± SEM of 3 replicates each. *P<0.05, VC = Vehicle Control (DMSO)] |
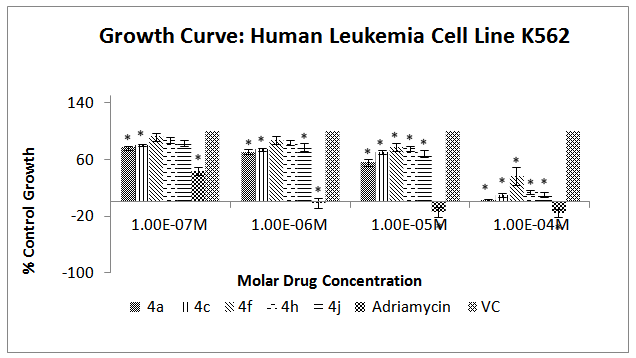 | Figure 5. Effect of different chalcone derivatives on the percentage growth inhibition of the K562 cell line[Values are Mean ± SEM of 3 replicates each. *P<0.05, VC = Vehicle Control (DMSO)] |
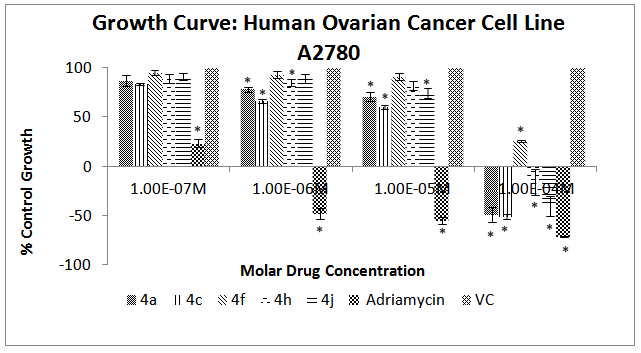 | Figure 6. Effect of different chalcone derivatives on the percentage growth inhibition of the A2780 cell line[Values are Mean ± SEM of 3 replicates each. *P<0.05, VC = Vehicle Control (DMSO)] |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
3.2. Chemistry and Cytotoxic Activity
- Although tubulin binding ability has been the most thoroughly investigated mode of action of chalcones, a number of other diverse mechanisms have been proposed that may explain their potent cytotoxic activity in diverse cell lines. Some of these include TRAIL-mediated as well as hypoxia-induced apoptosis, mitochondrial membrane disruption, inhibition of cell signal transduction as well as NF-κB (Nuclear Factor kappa-light chain-enhancer of activated B cells) pathways, and cell cycle arrest[27].The general trend in the correlation of cytotoxicity with different A-ring substituents pointed to a net decrease in biological activity. Typically, the parent chalcone 4a was found to have a higher cytotoxic activity in relation to the GI50 values compared to the substituted derivatives. This may point to the pharmacological significance of an intact A-ring, but other studies[28] have reported higher cytotoxic activity of different A-ring substitutions compared to the unsubstituted compound. This leads us to the reasonable assessment that it is not the substitutions per se but the regiochemistry of substitution that has a profound impact on the biological activity. Another interesting observation that has been evident all through the study is the characteristic biphasic response (growth hormesis) of this class of compounds that has previously been reported[26, 29-31]. Many hypotheses have been propounded to shed light on this phenomenon. First, the proliferation control mechanisms in the cancer cell might respond to the low growth inhibiting concentration of chalcone by inducing regulatory corrections which might be disabled at higher concentrations leading to apoptosis[32]. Second, it is known that chalcones and other flavonoid moieties have a dynamic equilibrium between the anti-oxidant and pro-oxidant properties that is concentration dependent. For the same reason, it can be concluded that a higher cellular chalcone concentration might contribute to oxidative reactions leading to build up of reactive oxygen species (ROS), which in turn induce cytotoxic response in cell cultures. However at lower concentrations, chalcones might act as anti-oxidants and protect the cells from oxidative damage[32]. Finally, the compounds might act as cell cycle specific modulators depending on the cellular concentrations and target the S phase at lower and G1 phase at higher concentrations[29, 33]. The pro-oxidant activity of the flavonoid family of natural products including their biosynthetic precursor compounds in the form of chalcones has been studied in detail. Strikingly, one of the major mechanisms of cytotoxicity of these moieties at higher concentrations has been attributed to their pro-oxidant action[34, 35]. Furthermore, the catalyzing activity of endogenous peroxidases naturally present in cell cultures at higher concentrations may also trigger high ROS levels leading to oxidative stress, mitochondrial dysfunction and cell death[36, 37]. The work reported herein attempts to establish structure function correlations with regard to cytotoxicity and as such, a mechanistic investigation of these derivatives is out of scope of this study. However, given our observation of biphasic response in all the tested analogs coupled with the literature precedence, there is significant clarity with regard to the existence of a pro-oxidant machinery effecting cytotoxicity in cancer cells. Therefore, it can be reasonably hypothesized that the cytotoxic activity of these compounds is likely to arise at least in part due to their pro-oxidant activity. From among the analogs evaluated, 4c bearing the 2’-methyl substituent was found to have comparable activity to the parent 4a. In this study, the 3’-nitro as well as the 4’-methoxy (4f and 4h respectively) derivatives were the worst affected in terms of their cytotoxic activity due to the substitutions on A-ring. Significantly, our results corroborate previous findings relating to the suppression of the cytotoxic effects due to the 4’-methoxy substitution on MCF-7 cell line[38]. The higher cytotoxicity of the unsubstituted chalcone compared to the different derivatives with substitutions on the A-ring alone as well as on both the rings has also been reported earlier. In another study, it was observed that the cytotoxicity of the 4’-methoxy derivative was drastically inhibited when chlorine was introduced at the ortho position of the A-ring, while there was a significant increase in the activity when a methyl group was introduced at the same position. The activity was again found to significantly increase by placing a hydroxy substituent at the ortho position in the A-ring[38]. These data clearly indicate that it is not only the nature and position of the substituent(s) on any particular ring, but also the interplay of different substitutions on other rings that can affect the overall biological activity of any compound[38]. Therefore, derivatives that are not significantly active in the current study may have greatly altered activity when combined with other substituents possessing characteristic electronic and steric properties.
4. Conclusions
- In summary, we rationally designed and readily synthesized a set of monosubstituted chalcone derivatives and evaluated them for potential cytotoxic activity against five human cancer cell lines. Although all the compounds under investigation exhibited lesser GI50 values than adriamycin, some interesting observations could be made from the results. The unsubstituted parent chalcone 4a was found to show higher cytotoxic activity than most of the substituted derivatives. In general, the substitutions at 2’- (ortho-), 3’- (meta-) or 4’- (para-) positions lead to a suppression of the cytotoxic activity. Among the substituted congeners investigated, the 2’-methyl derivative 4c was found to exhibit the most potent cytotoxic activity. This study has thus provided valuable preliminary information on the effects of various substituents on the cytotoxic profile of chalcones. This in turn may act as an impetus for the design of more potent chalcone derivatives as anticancer leads. Further studies with larger libraries of compounds screened against diverse cell lines are needed to establish a strong and reliable antiproliferative SAR profile for these molecules.
ACKNOWLEDGEMENTS
- The authors would like to express their gratitude to Dr. R. Gude at Advanced centre for Treatment, Research and Education in Cancer for his advice and scientific evaluation of the manuscript. The authors wish to thank the scientific assistants at Anti-Cancer Drug Screening Facility of Advanced centre for Treatment, Research and Education in Cancer for their support in carrying out the cytotoxicity assay.
References
| [1] | Rahman M. A., 2011, Chalcone: A valuable insight into the recent advances and potential pharmacological activities., Chem. Sci. J., 2011, CSJ-29. |
| [2] | Awasthi, S. K., Mishra, N., Kumar, B., Sharma, M., Bhattacharya, A., Mishra, L. C., and Bhasin, V. K., 2009, Potent antimalarial activity of newly synthesized substituted chalcone analogues in vitro., Med. Chem. Res., 18(17), 407–420. |
| [3] | Cheng, M. S., Shili, R., and Kenyon, G. A., 2000, Solid phase synthesis of chalcones by Claisen-Schmidt condensations., Chin. Chem. Lett., 11(10), 851–854. |
| [4] | Lim, S. S., Kim, H. S., and Lee, D. U., 2007, In vitro antimalarial activity of flavonoids and chalcones., Bull. Korean Chem. Soc., 28(12), 2495–2497. |
| [5] | Achanta, G., Modzelewska, A., Feng, L., Khan, S. R., and Huang, P., 2006, A boronic-chalcone derivative exhibits potent anticancer activity through inhibition of the proteasome., Mol. Pharmacol., 70(1), 426–433. |
| [6] | Lunardi, F., Guzela, M., Rodrigues, A. T., Corre, R., Eger-Mangrich, I., Steindel, M., Grisard, E. C., Assreuy, J., Calixto, J. B., and Santos, A. R. S., 2003, Trypanocidal and leishmanicidal properties of substitution-containingchalcones., Antimicrob. Agents Chemother., 47(4), 1449–1451. |
| [7] | Yadav, H. L., Gupta, P., Pawar, P. S., Singour, P. K., and Patil, U. K., 2011, Synthesis and biological evaluation of anti- inflammatory activity of 1,3-diphenyl propenone derivatives., Med. Chem. Res., 20(4), 461–465. |
| [8] | Hamdi, N., Fischmeister, C., Puerta, M. C., and Valerga, P., 2011, A rapid access to new coumarinyl chalcone and substituted chromeno[4,3-c]pyrazol-4(1H)-ones and their antibacterial and DPPH radical scavenging activities., Med. Chem. Res., 20(4), 522–530. |
| [9] | Awasthi, S. K., Mishra, N., Dixit, S. K., Singh, A., Yadav, M., Yadav, S. S., and Rathaur, S., 2009, Antifilarial activity of 1,3-diarylpropen-1-one: Effect on glutathione-S-transferase, a phase-II detoxification enzyme., Am. J. Trop. Med. Hyg., 80(5), 764–768. |
| [10] | Bag, S., and Ramar, S., 2009, Synthesis and biological evaluation of α,β-unsaturated ketone as potential antifungal agents., Med. Chem. Res., 18(4), 309–316. |
| [11] | Yayli, N., Ucuncu, O., Yasar, A., Kucuk, M., Akyuz, E., and Karaoglu, S. A., 2006, Synthesis and biological activities of N-alkyl derivatives of o-, m-, and p-nitro (E)-4-azachalcones and stereoselective photochemistry in solution with theoretical calculations., Turk. J. Chem., 30(4), 505–514. |
| [12] | Kaushik, S., Kumar, N., and Drabu, S., 2010, Synthesis and anticonvulsant activities of phenoxychalcones., T. Ph. Res., 3, 257–262. |
| [13] | Zarghi, A., Zebardast, T., Hakimion, F., Shirazi, F. H., Rao, P. N. P., and Knaus E. E., 2006, Synthesis and biological evaluation of 1,3-diphenylprop-2-en-1-ones possessing a methanesulfonamido or an azido pharmacophore as cyclooxygenase-1/-2 inhibitors., Bioorg. Med. Chem., 14(20), 7044–7050. |
| [14] | Ducki, S., 2007, The development of chalcones as promising anticancer agents., IDrugs, 10(1), 42–46. |
| [15] | Lawrence, N. J., and McGown, A. T., 2005, The chemistry and biology of antimitotic chalcones and related enone systems., Curr. Pharm. Des., 11(13), 1679–1693. |
| [16] | Nowakowska, Z., 2007, A review of anti-infective and anti-inflammatory chalcones., Eur. J. Med. Chem., 42(2), 125–137. |
| [17] | Ducki, S., 2009, Antimitotic chalcones and related compounds as inhibitors of tubulin assembly., Anticancer Agents Med. Chem., 9(3), 336–347. |
| [18] | Peyrot, V., Leynadier, D., Sarrazin, M., Briand, C., Rodriquez, A., Nieto, J. M., and Andreu, J. M., 1989, Interaction of tubulin and cellular microtubules with the new antitumor drug MDL 27048 - a powerful and reversible microtubule inhibitor., J. Biol. Chem., 264(35), 21296 – 21301. |
| [19] | Ducki, S., Rennison, D., Woo, M., Kendall, A., Chabert, J. F. D., McGown, A. T., and Lawrence, N. J., 2009, Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1: Synthesis and biological evaluation of antivascular activity., Bioorg. Med. Chem., 17(22), 7698–7710. |
| [20] | Boumendjel, A., Boccard, J., Carrupt, P. A., Nicolle, E., Blanc, M., Geze, A., Choisnard, L., Wouessidjewe, D., Matera, E. L., and Dumontet, C., 2008, Antimitotic and antiproliferative activities of chalcones: Forward structure -activity relationship., J. Med. Chem., 51(7), 2307–2310. |
| [21] | Szliszka, E., Czuba, Z. P., Mazur, B., Sedek, L., Paradysz, A., and Krol, W., 2010, Chalcones enhance TRAIL-induced apoptosis in prostate cancer cells., Int. J. Mol. Sci., 11(1), 1–13. |
| [22] | Selassie, C. D., Shusterman, A. J., Kapur, S., Verma, R. P., Zhang, L., and Hansch, C., 1999, On the toxicity of phenols to fast growing cells. A QSAR model for a radical-based toxicity., J. Chem. Soc., Perkin Trans. 2, 1999(12), 2729–2733. |
| [23] | Sabzevari, O., Galati, G., Moridani, M. Y., Siraki, A., and O'Brien, P. J., 2004, Molecular cytotoxic mechanisms of anticancer hydroxychalcones., Chem. Biol. Interact., 148 (1-2), 57–67. |
| [24] | Ducki, S., Forrest, R., Hadfield, J. A., Kendall, A., Lawrence, N. J., McGown, A. T., and Rennison, D., 1998, Potentantimitotic and cell growth inhibitory properties of substituted chalcones., Bioorg. Med. Chem. Lett., 8(9), 1051–1056. |
| [25] | Vichai, V., and Kirtikara, K., 2006, Sulforhodamine B colorimetric assay for cytotoxicity screening., Nat. Protoc., 1(3), 1112–1116. |
| [26] | Hijova, E., 2006, Bioavailability of chalcones., Bratisl. Lek. Listy, 107(3), 80–84. |
| [27] | Zhang, E. H., Wang, R. F., Guo, S. Z., and Liu, B., 2013, An update on antitumor activity of naturally occurring chalcones., J. Evid. Based Complement. Altern. Med., 2013, 1–22. |
| [28] | Shibata, S., 1994, Anti-tumorigenic chalcones., Stem Cells, 12(1), 44–45. |
| [29] | Araújo, J. R., Gonçalves, P., and Martel, F., 2011, Chemopreventive effect of dietary polyphenols in colorectal cancer cell lines., Nutr. Res., 31(2), 77–87. |
| [30] | [30] Padhye, S., Ahmad, A., Oswal, N., Dandawate, P., Rub, R. A., Deshpande, J., Swamy, K. V., and Sarkar, F. H., 2010, Fluorinated 2’-hydroxychalcones as garcinol analogs with enhanced antioxidant and anticancer activities., Bioorg. Med. Chem. Lett., 20(19), 5818–5821. |
| [31] | Snijman, P. W., Swanevelder, S., Joubert, E., Green, I. R., and Gelderblom, W. C. A., 2007, The antimutagenic activity of the major flavonoids of rooibos (Aspalathus linearis): Some dose-response effects on mutagen activation-flavonoid interactions., Mutat. Res., 631(2), 111–123. |
| [32] | Van der Woude, H., Gliszczynska-Swiglo, A., Struijs, K., Smeets, A., Alink, G. M., and Rietjens, I. M. C. M., 2003, Biphasic modulation of cell proliferation by quercetin at concentrations physiologically relevant in humans., Cancer Lett., 200(1), 41–47. |
| [33] | Van Erk, M. J., Roepman, P., Van der Lende, T. R., Stierum, R. H., Aarts, J. M., Van Bladeren, P. J., and Van Ommen, B., 2005, Integrated assessment by multiple gene expression analysis of quercetin bioactivity on anticancer-related mechanisms in colon cancer cells in vitro., Eur. J. Nutr., 44(3), 143–156. |
| [34] | Aruoma, O., 2003, Methodological considerations for characterizing potential antioxidant actions of bioactive components in plant foods., Mutat. Res., 523, 9–20. |
| [35] | Galati, G., and O’Brien, P. J., 2004, Potential toxicity of flavonoids and other dietary phenolics: significance for their chemopreventive and anticancer properties., Free Radic. Biol. Med., 37(3), 287–303. |
| [36] | Azmi, A. S., Bhat, S. H., Hanif, S., and Hadi, S. M., 2006, Plant polyphenols mobilize endogenous copper in human peripheral lymphocytes leading to oxidative DNA breakage: a putative mechanism for anticancer properties., FEBS Lett., 580(2), 533–538. |
| [37] | Lee, K. W., Hur, H. J., Lee, H. J., and Lee, C. Y., 2005, Antiproliferative effects of dietary phenolic substances and hydrogen peroxide., J. Agric. Food Chem., 53(6), 1990–1995. |
| [38] | Syam, S., Abdelwahab, S. I., Al-Mamary, M. A., and Mohan, S., 2012, Synthesis of chalcones with anticancer activities., Molecules, 17(6), 6179–6195. |