-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Materials Engineering
p-ISSN: 2166-5389 e-ISSN: 2166-5400
2016; 6(4): 126-133
doi:10.5923/j.ijme.20160604.02

Electrochemical Study of the Oxidation on Ammonia and Cyanide by Cyclic Voltammetry
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLChao-Lang Kao1, Ching-wei Chang1, Po-Yu Hu2
1Department of Chemical and Materials Engineering, National Chin-Yi University of Technology, Taiwan
2Division of Water Technology Research, Material and Chemical Research Laboratories, Industrial Technology Research Institute, Taiwan
Correspondence to: Chao-Lang Kao, Department of Chemical and Materials Engineering, National Chin-Yi University of Technology, Taiwan.
| Email: |  |
Copyright © 2016 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

In the cyclic voltammetry (CV) of ammonia, no oxidation peak was observed before reaching the oxygen evolution potential (OEP) when using two types of dimensionally stable anode, Pt/Ti and IrO2/Ti. This suggests that the direct electrochemical removal of ammonia is extremely ineffective. The OEP of Pt/Ti (1.47 V) is higher than that of IrO2/Ti (1.25 V), and thus, the current efficiency (CE) of Pt/Ti is better than that of IrO2/Ti in removing ammonia, there were 5.18% and 4.79%, respectively. According to the CV curve of cyanide removal, both Pt/Ti and IrO2/Ti anodes presented significant oxidation peaks before reaching OEP. Therefore, the CE of direct electrochemical oxidation in removing cyanide is much better than that in removing ammonia, where the CE of Pt/Ti is better than that of IrO2/Ti, there were 22.09% and 17.18%, respectively. In the CV of sodium chloride, the chlorine evolution potential (CEP, 1.07 V) of the Pt/Ti anode was much lower than the OEP (1.47 V) of this anode. In contrast, the CEP of the IrO2/Ti anode (1.07 V) was similar to the OEP (1.25 V) of this anode. The chlorine evolution efficiency of the Pt/Ti anode was better than that of the IrO2/Ti anode, and thus, the indirect oxidation CE of the Pt/Ti anode was better than that of the IrO2/Ti anode for the removal of ammonia, there were 59.12 and 22.46, respectively. Similarly, the indirect oxidation CE of the Pt/Ti anode was better than that of the IrO2/Ti anode for the removal of cyanide, there were 92.78 and 40.34, respectively. When adding (NH4)2SO4 or NaCN to a NaCl electrolyte solution, the integral of the chlorine evolution/reduction peak concurrently decreased with increases in additive concentrations; the magnitude of this decrease for the Pt/Ti anode was greater than that for the IrO2/Ti anode, and the magnitude of the decrease when adding NaCN was greater than that when adding (NH4)2SO4. Changes in the integral reduction magnitude of reduction peaks imply that the CE of the Pt/Ti anode was higher than that of the IrO2/Ti anode for the removal of ammonia and cyanide, where the removal of cyanide was easier than that of ammonia.
Keywords: Cyclic Voltammetry, Dimensionally Stable Anode, Ammonia, Cyanide
Cite this paper: Chao-Lang Kao, Ching-wei Chang, Po-Yu Hu, Electrochemical Study of the Oxidation on Ammonia and Cyanide by Cyclic Voltammetry, International Journal of Materials Engineering , Vol. 6 No. 4, 2016, pp. 126-133. doi: 10.5923/j.ijme.20160604.02.
Article Outline
1. Background
- Pollution problems are becoming increasingly severe with the rapid development of technology. Many industrial processes generate substances that are hazardous to life and the environment. These substances must be appropriately treated in subsequent stages. In recent years, the application of electrochemical techniques to remove pollutants from water has been extensively researched because of the low cost, high material removal rate, and environmentally friendliness of these techniques.Electrochemical treatment typically uses direct and indirect oxidation of electrode surfaces to treat wastewater. Direct oxidation refers to the use of hydroxyl radical degradation, whereas indirect oxidation refers to the preparation of different oxidizing agents such as active chlorine, hydrogen peroxide [1], ozone [2], and persulfate [3], among which the preparation of active chlorine through electrochemical oxidation is a common method because of the abundance of chloride in the natural environment and in industrial wastewater [4]. Although active chlorine is prone to produce byproducts such as chlorates and perchlorates during degradation, the production of byproducts can be suppressed by selecting suitable electrode materials, changing current density, and controlling pH levels [5].Use of appropriate electrode materials to enhance current efficiency (CE) and reduce energy consumption is an essential aspect of consideration during degradation. Dimensionally stable anode (DSA) materials such as Pt/Ti and IrO2/Ti typically follow sequential fundamental steps of (1) adsorption, (2) surface diffusion, (3) chemical recombination (bond breakage, bond formation, and molecular recombination), and (4) desorption during electrocatalysis. Therefore, heterogeneous catalysis is governed by multiple surface parameters. Among these parameters, the crystal structure, surface composition, and electronic structure of a catalyst are the key reaction determinants [6]. Use of dimensionally stable anodes (DSAs) to produce chlorine gas and sodium hypochlorite is a common practice in the chloralkali industry. This method yields exceptional results in wastewater treatment because of excellent electrocatalytic properties, stability, and service life of DSA materials [7].
2. Methods
- Cyclic voltammetry (CV) was performed in a tripolar, single-compartment electrolytic cell using BioLogic SP-150. Working electrodes were made of Pt/Ti and IrO2/Ti (Kojin Co., Taiwan; work area = 0.785 cm2), counter electrodes were made of Pt, and reference electrodes were made of saturated calomel (MSE; 0.64 vs SHE). The scanning rate was set as 30 mv/s, and tests were conducted at room temperature. Prior to the tests, the working electrodes were cleaned using deionized water. They were then air-dried, and subsequently, NaCl, (NH4)2SO4, and NaCN electrolyte solutions were applied. The electrochemical characteristics of the working electrodes were concurrently assessed based on peak potential/current variance with a change in the scanning rate to deduce their reaction mechanisms.The electrochemical oxidation of ammonia and cyanide was performed in a 500-ml, single-compartment electrolytic cell. Anodes were made of Pt/Ti and IrO2/Ti, and cathodes were made of Pt/Ti. The working area was 5 × 10 cm2 with an anode-to-cathode area ratio of 2:1. A magnetic stirrer operating at 400 rpm was used to create convection, and tests were conducted at room temperature. The ammonia solution was prepared using (NH4)2SO4, which served as 2800 mg/L of NH3–N-simulated wastewater. The cyanide solution was prepared using NaCN, which served as 3600 mg/L of CN−-simulated wastewater. In addition, 0.6 g/L of sodium chloride was added as the oxidation catalyst for indirect oxidation. The efficiency of this indirect oxidation was then compared to that of direct oxidation. pH was controlled at target levels using NaOH or H2SO4. All solutions were prepared using ultra-pure water.A spectrophotometer (Hach Lange spectrophotometer; DR 900) was used to measure the concentrations of ammonia and cyanide. Nitrogen–ammonia reagent set, high-range Test N TubeTM AmVerTM 2606945 0.4-50.0 mg/L NH3-N and the cyanide reagent powder pillow 2106869, 2106969, 210069 0.002-0.240 mg/L CN− were used for testing.
3. Results
3.1. Direct Electrocatalytic Oxidation
- Many studies interpreted the adsorption behavior of oxygen on the surface of platinum using the Langmuir Model, where only a single layer of oxygen atoms adhered to the surface of platinum rather than multiple layers [8]. During the electrolysis of halogen-free solutions, oxygen atoms or hydroxyl radicals are adsorbed onto the anode surface. Thus, the adsorption capacity of oxygen atoms or hydroxyl radicals differs depending on the anode material. Free radicals with strong adsorption are prone to be adsorbed; thus, an adequate density of oxygen atoms will be adsorbed onto the anode surface, which increases the covalent precipitation of oxygen and reduces the oxygen evolution potential (OEP) of anodes (e.g., IrO2/Ti electrodes). Moreover, free radicals are less likely to desorb because of their strong adherence to the anode surface. Thus, anode materials with low OEP typically demonstrate weak oxidation potential. In contrast, anode materials with poor oxygen atom adsorption (such as Pt/Ti electrodes) can relatively more easily desorb oxygen atoms or hydroxyl radicals from the anode surface, thus demonstrating a relatively stronger oxidation potential. Such electrodes require a high electric potential to enable the anode surface to adsorb an adequate density of oxygen atoms to precipitate oxygen, suggesting that such anode materials possess high OEP. In summary, anode materials with low OEP typically demonstrate weak direct electrocatalytic oxidation. In contrast, anode materials with high OEP demonstrate strong direct electrocatalytic oxidation.CE can be calculated using Eq. (1), where C0 and C represent the molarity of consumption, n represents the valence number, V represents the solution volume, F represents the Faraday constant, I represents current, and t represents time.
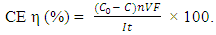 | (1) |
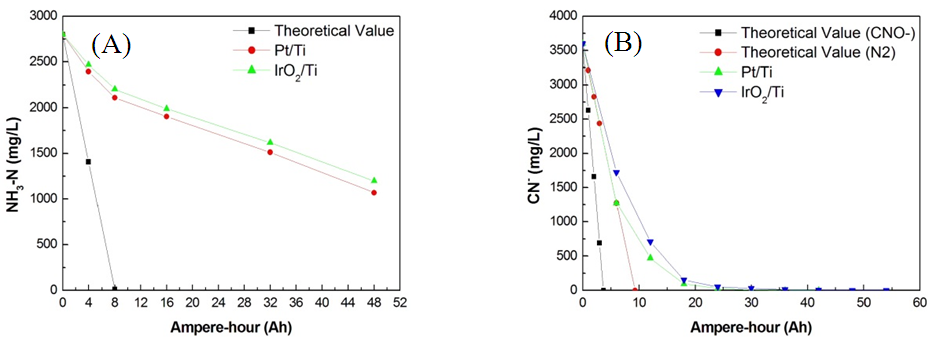 | Figure 1. Direct electrochemical oxidation in (A) 2800 mg/L of NH3–N and (B) 3600 mg/L of CN− with Pt/Ti and IrO2/Ti anodes and Pt/Ti cathodes (pH = 8.5; current = 2 A; and rotation = 400 rpm) |
3.2. Indirect Electrocatalytic Oxidation
- During indirect electrocatalytic oxidation, NaCl was added as the oxidation catalyst. Thus, indirect electrocatalytic oxidation reactions are measured by the efficiency of chlorine evolution. Cl− is oxidized on an anode to produce Cl2. Then, Cl2 is dissolved in water to form HClO or ClO-. This active chlorine reacts with ammonia and cyanide to form nitrogen, thus achieving removal. The reaction process can be calculated using Eqs. (2)–(4). CV was used to investigate the OEP and chlorine evolution potential (CEP) of the Pt/Ti and IrO2/Ti anodes and the integrated area size of the reduction characteristic peaks, both of which can be used to elaborate the indirect oxidation mechanisms of ammonia and cyanide.
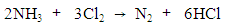 | (2) |
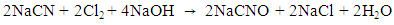 | (3) |
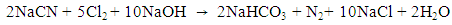 | (4) |
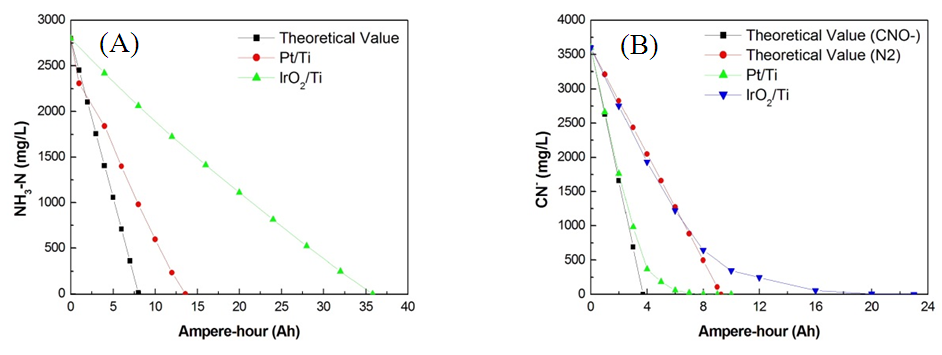 | Figure 2. Indirect electrochemical oxidation in (A) 2800 mg/L of NH3–N and (B) 3600 mg/L of CN− with Pt/Ti and IrO2/Ti anodes and Pt/Ti cathodes (NaCl additive = 0.6 g/L; pH = 8.5; current = 2 A; and rotation = 400 rpm) |
 | Figure 3. CV spectra of (A) Pt/Ti anode and (B) IrO2/Ti anode with 1 M H2SO4, Na2SO4, NaOH, and NaCl |
3.3. Using CV to Examine Ammonia and Cyanide Removal Potential of Direct Electrocatalytic Oxidation
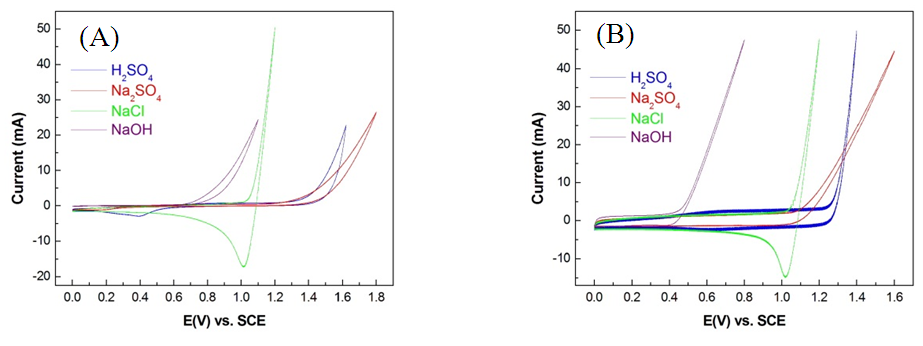
- The CV oxygen evolution curve (Fig. 4A) shows that the OEP of the Pt/Ti anode (1.47V) was higher than that of the IrO2/Ti anode (1.25V). As previously discussed, anode materials with high OEPs typically exhibit a strong direct electrocatalytic oxidation potential. Therefore, the CE of electrolytic oxidation using the Pt/Ti anode was higher than that using the IrO2/Ti anode. No ammonia oxidation peak was observed in CV test results prior to the OEP of both Pt/Ti and IrO2/Ti anodes, as shown in Figs. 4B and 4C. These results suggest that the oxidation decomposition potential (ODP) of ammonia was higher than its OEP and that the current was largely consumed during oxygen precipitation, limiting the direct electrolytic oxidation removal efficiency of the anodes. Moreover, CE drastically decreased with the reduction of NH3-N concentrations, suggesting that the effective use of direct electrocatalytic oxidation removal in wastewater with low concentrations of ammonia is extremely difficult [9]. In contrast, cyanide oxidation curves were observed in CV test results prior to the OEP of both Pt/Ti and IrO2/Ti anodes, as shown in Fig. 5; this was particularly evident for the Pt/Ti anode. These results verify that cyanide can be easily removed through the direct oxidation of the anodes and that the ODP of cyanide is lower than that of ammonia. That is, cyanide is more effectively decomposed through oxidation compared with ammonia. In summary, the removal of cyanide via direct electrocatalytic oxidation is feasible.
 | Figure 4. CV spectral diagrams of (A) Pt/Ti and IrO2/Ti anodes in 1 M Na2SO4 solution; (B) Pt/Ti anode in 1 M (NH4)2SO4 solution; and (C) IrO2/Ti anode in 1 M (NH4)2SO4 solution |
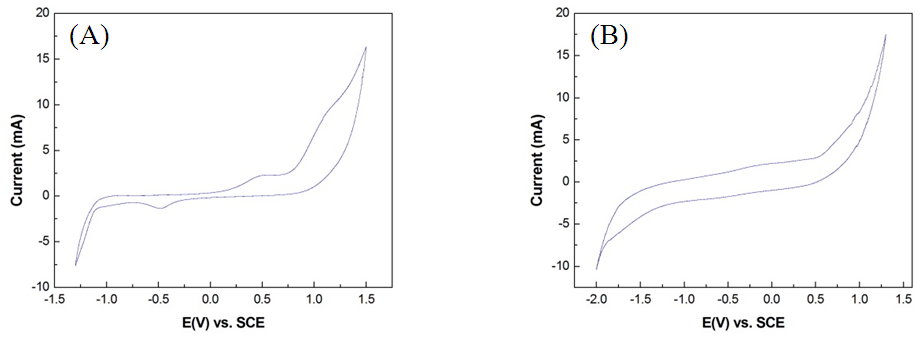 | Figure 5. CV spectral diagrams of (A) Pt/Ti anode and (B) IrO2/Ti anode in 1 M NaCN + 1 M NaOH solution |
3.4. Using CV to Examine Reduction Peaks of Chlorine Evolution
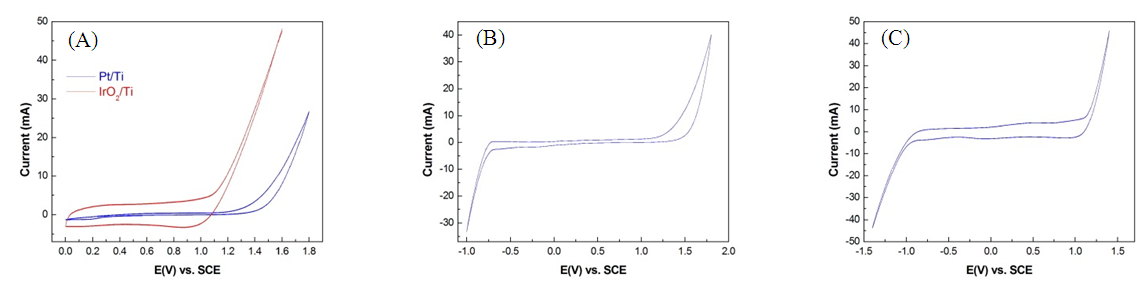
- In the NaCl electrolyte solution, Cl− on the anode surface lost electrons and oxidized into active chlorine atoms before the anode potential reached CEP. These electrons adhered onto the anode surface. Chlorine has the highest electron affinity among all elements (349 KJ/mol). Thus, as the anode potential gradually reduces, chlorine strongly recaptures electrons and returns to Cl−. When there is no substance in the electrolyte solution for chlorine atoms to oxidize into electrons, chlorine atoms can only recapture electrons from an anode; this instantaneously causes the electron flow of the anode to reverse into a reduction current, forming a particular reduction peak. This phenomenon has been mentioned in numerous previous studies [10, 11]. This reduction peak is evident in halogen elements with high electron affinity, as shown in Fig. 6A and B. However, reduction peaks were not observed during the oxygen evolution of the Pt/Ti and IrO2/Ti anodes because oxygen atoms have a low electron affinity level (141 KJ/mol).
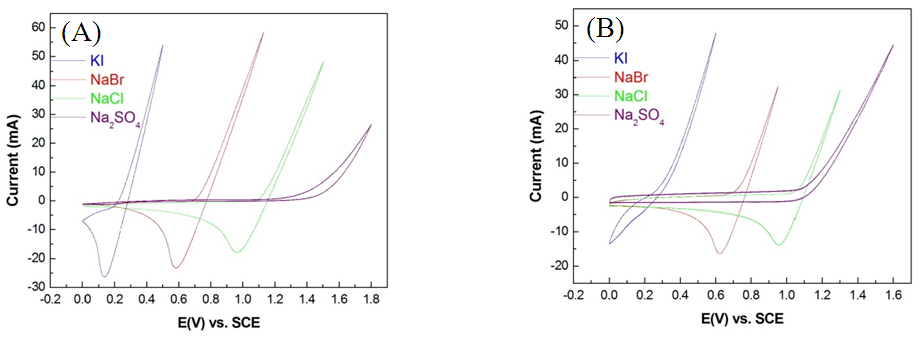 | Figure 6. CV spectra of (A) Pt/Ti anode and (B) IrO2/Ti anode with 1 M KI, NaBr, NaCl, and Na2SO4 |
3.4.1. Effects of Adding (NH4)2SO4 to NaCl Electrolyte Solution of Anodes on Integral of Reduction Peaks Observed in CV Results
- The magnitude of the integral of a reduction peak corresponded to the amount of chlorine atoms adsorbed onto the anode surface. When 0.1 M and 1 M of (NH4)2SO4 were added into 1 M of the NaCl electrolyte solution, chlorine atoms on the anode surface attracted the electrons of NH4+ and returned to Cl−. As the concentrations of NH4+ increased, reactions between chlorine atoms and NH4+ also increased, reducing the amount of electrons adsorbed by the anodes and the magnitude of the integral of the reduction peak.The present study compared three electrolyte solutions applied to the Pt/Ti and IrO2/Ti anodes, specifically (1) 1 M NaCl, (2) 1 M NaCl + 0.1 M (NH4)2SO4, and (3) 1 M NaCl + 1 M (NH4)2SO4. As illustrated in Fig. 7, the integral of reduction peaks observed in CV concurrently reduced with an increase in (NH4)2SO4 concentration. The integral of the reduction peak observed during the chlorine evolution of the Pt/Ti anode reduced from −108.631 mAs-1 (Fig. 8A) to −44.089 mAs-1 (Fig. 8B) at a reduction magnitude of 59.4%. It later reduced to −19.819 mAs-1 (Fig. 8C) at a total reduction magnitude of 81.8%. The integral of the reduction peak observed during the chlorine evolution of the IrO2/Ti anode reduced from −92.826 mAs-1 (Fig. 8D) to −55.963 mAs-1 (Fig. 8E) at a reduction magnitude of 39.7%. It later reduced to −43.177 mAs-1 (Fig. 8F) at a total reduction magnitude of 53.5%.
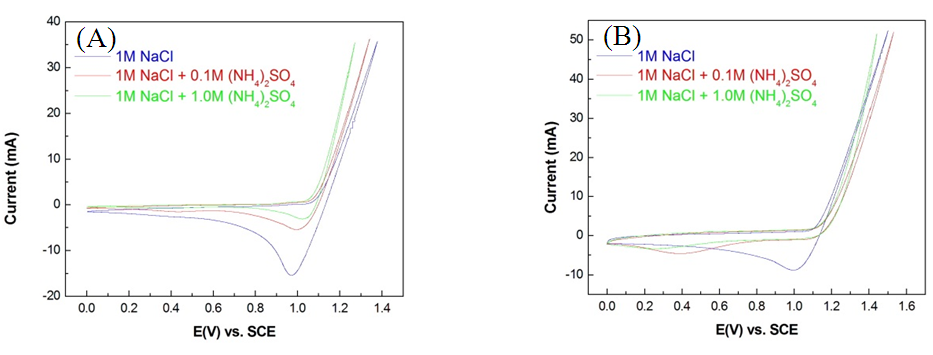 | Figure 7. CV spectral diagrams of (A) Pt/Ti anode and (B) IrO2/Ti anode in 1 M NaCl, 1 M NaCl + 0.1 M (NH4)2SO4, and 1 M NaCl + 1.0 M (NH4)2SO4 solutions |
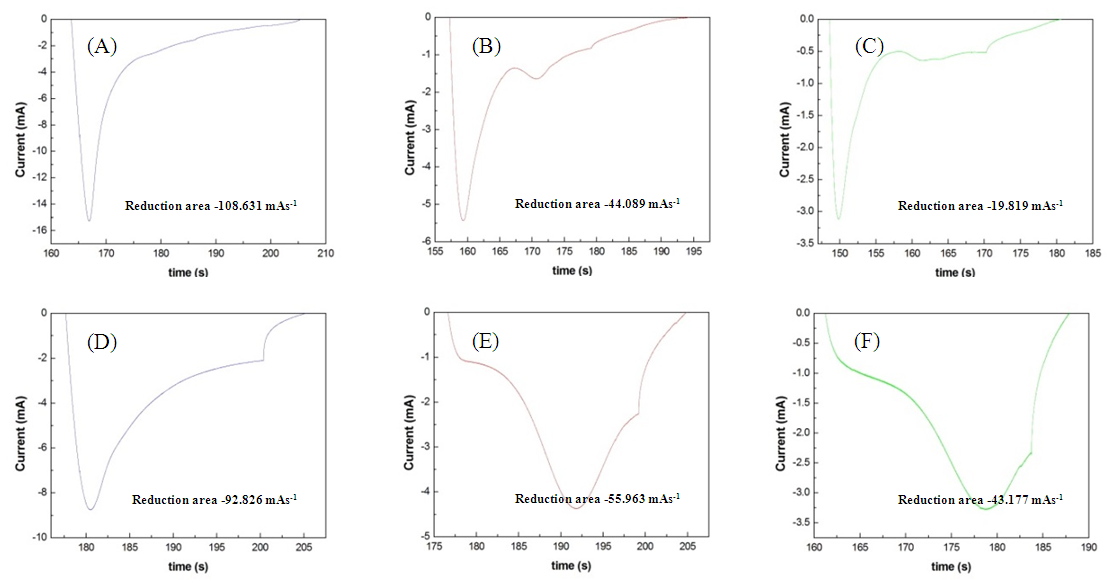 | Figure 8. Integral of reduction peak of Pt/Ti anode in (A) 1 M NaCl, (B) 1 M NaCl + 0.1 M (NH4)2SO4, and (C) 1 M NaCl + 1 M (NH4)2SO4 and that of IrO2/Ti anode in (D) 1 M NaCl, (E) 1 M NaCl + 0.1 M (NH4)2SO4, and (F) 1 M NaCl + 1 M (NH4)2SO4 |
3.4.2. Effects of Adding NaCN to NaCl Electrolyte Solution of Anodes on Integral of Reduction Peaks Observed in CV Results
- The present study compared three electrolyte solutions applied to the Pt/Ti and IrO2/Ti anodes, specifically (1) 1 M NaCl, (2) 1 M NaCl + 0.1 M (NH4)2SO4, and (3) 1 M NaCl + 1 M (NH4)2SO4. As illustrated in Fig. 9, the integral of reduction peaks observed in CV concurrently reduced with an increase in NaCN concentration. The integral of the reduction peak observed during the chlorine evolution of the Pt/Ti anode reduced from −119.106 mAs-1 (Fig. 10A) to −85.624 mAs-1 (Fig. 10B) at a reduction magnitude of 28.1%. It later reduced to −1.985 mAs-1 (Fig. 10C) at a total reduction magnitude of 98.3%. The integral of the reduction peak observed during the chlorine evolution of the IrO2/Ti anode reduced from −92.826 mAs-1 (Fig. 10D) to −21.387 mAs-1 (Fig. 10E) at a reduction magnitude of 77%. It later reduced to −14.013 mAs-1 (Fig. 10F) at a total reduction magnitude of 78.8%. The integral of the reduction peak concurrently decreased with an increase in NaCN concentration, where the Pt/Ti anode exhibited a greater reduction than the IrO2/Ti anode. The integral of the reduction peaks of both Pt/Ti and IrO2/Ti anodes exhibited a much greater reduction when NaCN was added to the electrolyte than when (NH4)2SO4 was added, suggesting that through oxidation, cyanide is easier to decompose than ammonia.
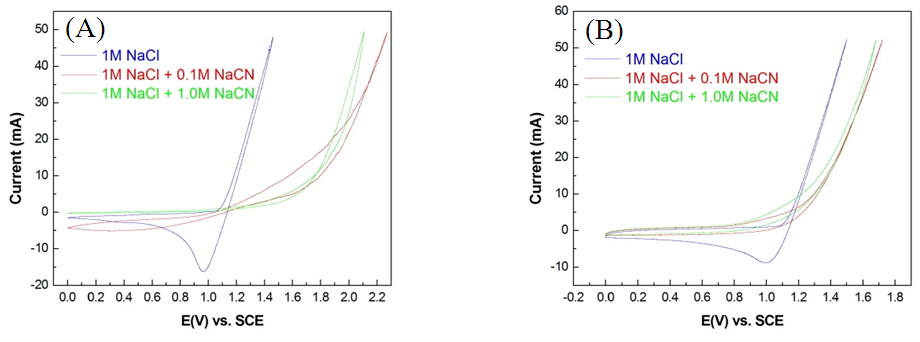 | Figure 9. CV spectral diagrams of (A) Pt/Ti anode and (B) IrO2/Ti anode in 1 M NaCl, 1 M NaCl + 0.1 M NaCN, and 1 M NaCl + 1.0 M NaCN solutions |
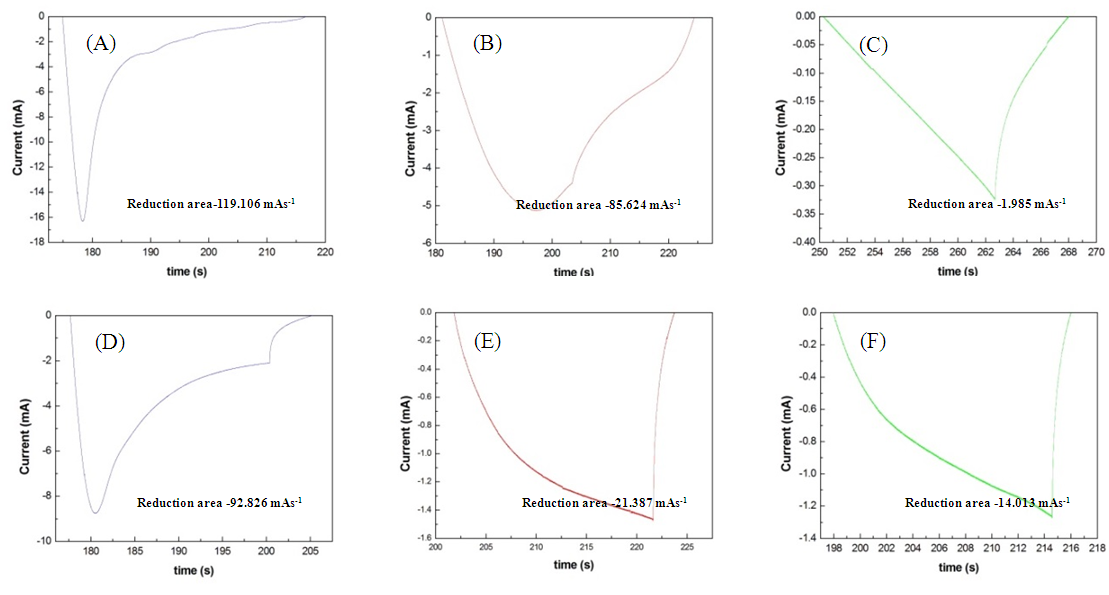 | Figure 10. Integral of reduction peak of Pt/Ti anode in (A) 1 M NaCl, (B) 1 M NaCl + 0.1 NaCN, and (C)1 M NaCl + 1 M NaCN and that of IrO2/Ti anode in (D) 1 M NaCl, (E) 1 M NaCl + 0.1 M NaCN, and (F) 1 M NaCl + 1 M NaCN |
3.5. Conclusions
- (1) CV curves for ammonia showed that no ammonia oxidation peak was observed in CV test results prior to the OEP of both Pt/Ti and IrO2/Ti anodes, suggesting that the ODP of ammonia was higher than its OEP and that the direct electrolytic oxidation removal efficiency of the anodes was limited. These results were verified by the electrolysis experiment results of ammonia.(2) CV curves for cyanide showed that significant cyanide oxidation peaks were observed in CV test results prior to the OEP of both Pt/Ti and IrO2/Ti anodes, suggesting that the ODP of cyanide was lower than that of ammonia. The electrolysis experiment results of cyanide showed that the cyanide removal CEs of both Pt/Ti and IrO2/Ti anodes were higher than those of ammonia removal CEs, suggesting that the removal of cyanide was easier than that of ammonia via oxidation. Moreover, the OEP of the Pt/Ti anode was higher than that of the IrO2/Ti anode. Therefore, in terms of the direct electrocatalytic oxidation reactions of the two anodes, the CE of the Pt/Ti anode was higher than that of the IrO2/Ti anode for the removal of both cyanide and ammonia.(3) CEPs of both Pt/Ti and IrO2/Ti anodes were lower than their OEPs. Among the two anodes, the CEP and OEP margins of the IrO2/Ti anode were smaller. Oxygen precipitation often occurs during electrolysis, hindering the CEP of the Pt/Ti anode. The results of the indirect electrocatalytic oxidation experiment to remove ammonia verified that the CE of the Pt/Ti anode was greater than that of the IrO2/Ti anode. The results of the same experiment to remove cyanide verified that the CE of the Pt/Ti anode was greater than that of the IrO2/Ti anode when NaCl was used as the oxidation catalyst for indirect electrocatalytic oxidation.(4) In the NaCl electrolyte solution, the magnitude of the integral of the reduction peak, as illustrated by the CV curve, corresponded to the amount of chlorine atoms adsorbed onto the anode surface. When (NH4)2SO4 was added to the NaCl electrolyte solution, the integral of the chlorine evolution/reduction peak of the Pt/Ti anode concurrently decreased with an increase in NaCN concentration, suggesting that the number of chlorine atoms adhered to the surface of the Pt/Ti anode decreased because they reverted to Cl− ions via the oxidation of NH4+ ions. The integral reduction magnitude of the reduction peak of the IrO2/Ti anode was less than that of the Pt/Ti anode. When NaCN was added to the NaCl electrolyte solution, the integral reduction magnitude of the reduction peak became relatively more evident. The change in the integral reduction magnitude of reduction peaks implies that the CE of the Pt/Ti anode in removing ammonia and cyanide was higher than that of the IrO2/Ti anode, where the removal of cyanide was easier than that of ammonia via oxidation.(5) Use of NaCl as the oxidation catalyst in electrolytic oxidation to remove pollutants from water is a common practice. When examining the removal of different pollutants, CV curves can help us decide the oxidation feasibility and electro-catalytic performance of an anode and various pollutants by observing OEP/CEP levels as well as changes in the integral magnitude of chlorine evolution/reduction peaks.