Saparbai Kalbaev1, Mashkhura Raximberdiyeva1, Lola Bakhriddinova1, Nuritdin Kattaev2, Khamdam Akbarov2
1PhD Student, Department of Physical Chemistry, National University of Uzbekistan, Tashkent, Uzbekistan
2DSc, Professor, Department of Physical Chemistry, National University of Uzbekistan, Tashkent, Uzbekistan
Correspondence to: Saparbai Kalbaev, PhD Student, Department of Physical Chemistry, National University of Uzbekistan, Tashkent, Uzbekistan.
| Email: |  |
Copyright © 2025 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Abstract
The thermal behavior of a semi-permeable cellulose membrane (AS) and its composites with polypyrrole (AS/PPy) and p-toluenesulfonic acid (AS/PPy–PTSA) was investigated by thermogravimetric (TGA) and differential thermal analysis (DTA) from 25 to 600°C. All samples decomposed in three stages, but the presence of conducting polymer and dopant strongly affected their stability. Pristine cellulose exhibited major degradation at 344.7°C with a total mass loss of 78% and a char yield of 22%. Incorporation of polypyrrole shifted the main decomposition to 302.8°C, reduced overall mass loss to 70%, and increased char yield to 30%. PTSA doping further lowered Tmax to 250.9°C but produced the highest char residue (~40%), reflecting the dual role of ionic doping. Kinetic analysis using the Kissinger method indicated a systematic decrease in activation energy from 53.9 to 43.8 kJ·mol⁻¹, while Coats–Redfern modeling suggested diffusion-limited processes. Thermodynamic parameters (positive ΔH, negative ΔG, high positive ΔS) confirmed endothermic but spontaneous and entropy-driven decomposition. The results demonstrate that while doping reduces onset stability, it enhances carbonization and residue stability, making AS/PPy–PTSA composites promising for supercapacitors, sensors, flexible electronics, and EMI shielding.
Keywords:
Cellulose, Polypyrrole, P-toluenesulfonic acid, Composite materials, Thermal analysis, TGA/DTA, Activation energy, Thermodynamics
Cite this paper: Saparbai Kalbaev, Mashkhura Raximberdiyeva, Lola Bakhriddinova, Nuritdin Kattaev, Khamdam Akbarov, Study of Non-Isothermal Degradation of Cellulose-Polythiophene Composites, International Journal of Materials and Chemistry, Vol. 15 No. 3, 2025, pp. 64-70. doi: 10.5923/j.ijmc.20251503.07.
1. Introduction
Conducting polymer composites have gained considerable attention due to their unique combination of electrical, mechanical, and functional properties. Cellulose, an abundant, renewable, and biodegradable biopolymer, is an attractive substrate for such composites thanks to its flexibility, porosity, and biocompatibility [1]. However, pristine cellulose exhibits low thermal stability and limited char-forming ability, restricting its direct use in electronics, energy storage, and protective applications [2].Polypyrrole (PPy) is one of the most extensively studied conducting polymers, offering high conductivity, environmental stability, and facile synthesis [3]. In situ oxidative polymerization of pyrrole on cellulose provides a simple and effective route to fabricate PPy/cellulose composites with strong interfacial adhesion and enhanced electrochemical performance. Such materials are widely explored in sensors, supercapacitors, antistatic coatings, and flexible electronics [4-5]. Yet, incorporating PPy alters the thermal decomposition behavior of cellulose, making systematic analysis of degradation pathways essential for practical use.Doping represents another powerful tool to tailor the performance of conducting polymers. p-Toluenesulfonic acid (PTSA) is a widely used dopant that enhances charge stabilization and conductivity in polypyrrole [6]. At the same time, PTSA can influence thermal behavior by lowering the decomposition onset while promoting carbonization and higher char yields. This dual effect highlights the importance of thermal studies to understand the trade-off between initial stability and residue stability [7].Thermogravimetric (TGA) and differential thermal analysis (DTA), combined with kinetic and thermodynamic modeling (Kissinger and Coats–Redfern methods), provide valuable insights into degradation stages, activation energies, enthalpic and entropic contributions, and residue formation [8]. Such information is critical to correlate structure with thermal performance and to design stable cellulose-based conductive composites [9-10].In this work, we investigate the thermal decomposition of cellulose (AS), cellulose/polypyrrole composite (AS/PPy), and its PTSA-doped analogue (AS/PPy–PTSA). By comparing decomposition profiles, kinetic parameters, and thermodynamic functions, we aim to clarify how PPy and PTSA modify the degradation mechanisms and to assess their potential for applications in supercapacitors, sensors, flexible electronics, and electromagnetic shielding.
2. Materials and Methods
Materials. Pyrrole (Py, ≥99%, freshly distilled before use), ferric chloride hexahydrate (FeCl₃·6H₂O, ≥99%), and p-toluenesulfonic acid monohydrate (PTSA·H₂O, ≥98%) were purchased from commercial suppliers and used as received unless otherwise noted. Microcrystalline semi-permeable cellulose (AS) was employed as the supporting substrate. Deionized water and analytical-grade ethanol were used throughout the experiments.Synthesis of AS/PPy composite. Cellulose membranes were immersed in 0.1 M pyrrole solution for 2 h. Oxidative polymerization was initiated by slow addition of 0.15 M FeCl₃ solution at a Py:Fe³⁺ ratio of 1:1.5, following procedures adapted from Stejskal and Gilbert [6]. Polymerization continued for 6 h under stirring. The black PPy-coated membranes were washed with water/ethanol and dried under vacuum at 50°C for 24 h.Preparation of AS/PPy–PTSA composite. AS/PPy samples were immersed in 0.1 M PTSA solution for 12 h, washed, and dried under vacuum. Sulfonate doping was expected to stabilize charge carriers, as reported for PTSA-doped PPy [7].Thermal analysis. Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) were performed using a simultaneous thermal analyzer (Netzsch STA or equivalent). Approximately 10 mg of each sample (AS, AS/PPy, AS/PPy–PTSA) was placed in alumina crucibles and heated from 25 to 600°C under an air atmosphere with a heating rate of 10°C·min⁻¹. Both mass loss (TG) and heat flow (DTA) curves were recorded.Kinetic and thermodynamic evaluation. Kinetic parameters were calculated from the TG/DTG data using the Kissinger method:ln(β/T²) = ln(AR/Ea) – Ea/RTwhere β is the heating rate, T is the temperature at the maximum decomposition rate, A is the pre-exponential factor, R is the gas constant, and Ea is the activation energy.The Coats–Redfern integral method was also applied to evaluate Ea under various mechanistic models:ln(g(α)/T²) = ln(AR/βEa) – Ea/RTwhere g(α) is the integral conversion function.Thermodynamic parameters including enthalpy of activation (ΔH), Gibbs free energy (ΔG), and entropy change (ΔS) were calculated from Ea values according to standard transition state theory equations.
3. Result and Discussion
Thermal decomposition of cellulose (AS)The thermal degradation of the pristine semi-permeable cellulose membrane (AS) was examined as a reference system (fig.1). Cellulose, as a natural polysaccharide composed of linear β-1,4-linked D-glucose units, contains a high density of hydroxyl groups that form a complex network of intra- and intermolecular hydrogen bonds. This structural feature provides rigidity and crystallinity but also makes cellulose relatively sensitive to thermal stress, because hydrogen bonding destabilizes at elevated temperatures and accelerates chain scission reactions.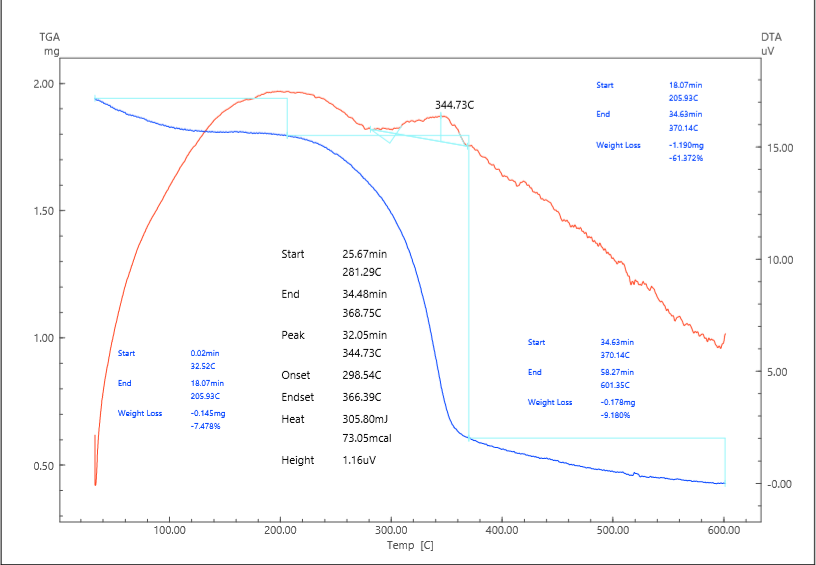 | Figure 1. Derivatogram of semipermeable cellulose (AS) |
The TGA/DTA curves of AS reveal a characteristic three-step degradation process. The first stage occurs between 32.5 and 205.9°C and is associated with the removal of physically adsorbed water and loosely bound volatile species. The mass loss of approximately 7.5% reflects the strong hygroscopic nature of cellulose, which is capable of retaining moisture within its amorphous domains and microcapillaries. The weak endothermic signal recorded in the DTA curve supports the interpretation that this step is governed by evaporation rather than chemical decomposition:H₂O(ads) → H₂O(gas).The second stage, occurring in the range of 205.9–370.1°C and corresponding to a major weight loss of about 61.4%, represents the primary thermal decomposition of the cellulose backbone. At this point, the cleavage of glycosidic bonds and depolymerization reactions dominate. The DTA trace shows a sharp exothermic peak at 344.7°C, which is consistent with the release of significant amounts of chemical energy as covalent bonds within the carbohydrate polymer are ruptured. The degradation pathway involves several concurrent processes. First, cellulose undergoes dehydration and depolymerization to form levoglucosan as the main volatile intermediate:(C₆H₁₀O₅)n → C₆H₁₀O₅ (levoglucosan) + H₂O + CO + CO₂.Secondary pathways include fragmentation of levoglucosan to produce furan derivatives such as furfural and hydroxymethylfurfural (HMF):C₆H₁₀O₅ → C₅H₄O₂ (furfural) + CH₂O,C₆H₁₀O₅ → C₆H₆O₃ (HMF) + 2 H₂O.The third stage of degradation occurs between 370.1 and 601.4°C, where an additional 9.2% weight loss is observed. This phase corresponds to the slow oxidation and burnout of the carbonaceous residue that forms after depolymerization. The process can be described by the oxidation of amorphous char into gaseous carbon dioxide:C(s) + O₂ → CO₂.The overall mass reduction reaches approximately 78%, leaving a final char yield of 22%. This low residual fraction highlights the organic nature of cellulose and its limited ability to form stable carbonaceous structures, which is a significant drawback when thermal stability or flame resistance is required.From a kinetic standpoint, the Kissinger method yielded an activation energy (Ea) of 53.9 kJ·mol⁻¹ based on the maximum decomposition temperature (344.7°C), which is consistent with previously reported values for native cellulose (50–60 kJ·mol⁻¹). The Coats–Redfern integral method provided Ea values in the range of 30–90 kJ·mol⁻¹ depending on the assumed reaction model, indicating that multiple mechanisms, including first-order chain scission and diffusion-controlled volatilization, operate simultaneously. The lowest activation energies correspond to one-dimensional diffusion models, confirming that the transport of volatile intermediates through the porous fibrous network governs the kinetics of the process.Thermodynamic analysis provides additional insights. The enthalpy of activation (ΔH) was determined to be 48.8 kJ·mol⁻¹, reflecting the energy barrier required to initiate bond cleavage. The Gibbs free energy (ΔG) of –55.6 kJ·mol⁻¹ demonstrates that the degradation process is thermodynamically favorable and spontaneous at the maximum decomposition temperature. The positive entropy change (ΔS ≈ +170 J·mol⁻¹·K⁻¹) indicates a transition toward greater molecular disorder, which is consistent with the transformation of an ordered solid phase into a complex mixture of gaseous products such as H₂O, CO, CO₂, furans, and small aldehydes.When compared with literature data, the behavior of AS is in excellent agreement with previous studies. For instance, similar three-step degradation patterns with Tmax between 330 and 350°C and char yields of 20–25% have been reported for a variety of cellulose sources, including microcrystalline cellulose and regenerated cellulose films.From a practical perspective, the results confirm that pure cellulose is unsuitable for applications requiring high thermal stability. The onset of significant mass loss around 300°C indicates that continuous operation at temperatures above 220–240°C would lead to irreversible degradation. Moreover, the low char yield reflects poor flame resistance, since the material lacks the ability to form a protective carbonaceous barrier.Thermal decomposition of AS/PPy compositeThe incorporation of polypyrrole (PPy) into the cellulose matrix significantly alters the decomposition behavior of the system, both in terms of thermal stability and the nature of the degradation products. Unlike pristine cellulose, which shows a sharp and well-defined exothermic event associated with polysaccharide depolymerization, the AS/PPy composite exhibits a more complex thermal profile arising from the coexistence of the carbohydrate substrate and the conducting polymer coating (fig.2).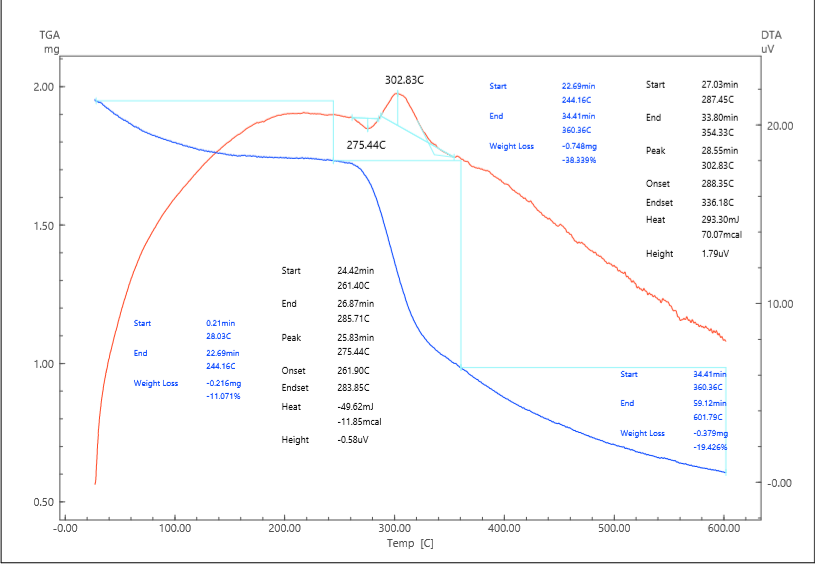 | Figure 2. Derivatogram of the AS/PPy composite |
The first stage of degradation (28.0–244.2°C, Δm = 11.1%) is more pronounced than in cellulose alone. This stage is associated with the desorption of water molecules retained by the highly porous and hydrophilic composite, as well as the removal of residual oxidants (e.g., FeCl₃-derived species) from the PPy synthesis. The broader temperature window for this event suggests stronger physical adsorption sites and a heterogeneous moisture distribution, consistent with the presence of both cellulose hydroxyl groups and PPy polar functionalities.The second stage (244.2–360.4°C, Δm = 38.3%) corresponds to the thermal decomposition of polypyrrole itself and partially overlaps with the degradation of cellulose. The DTA trace shows a strong exothermic peak at 302.8°C, which is attributed to the scission of PPy backbones. The decomposition mechanism involves cleavage of N–H and C–N bonds, leading to the release of ammonia and hydrogen cyanide:–NH– → NH₃–C–N– → HCN(C₄H₃N)n → n C₄H₃N + NH₃ + HCN + H₂OAt the same time, the cellulose fraction continues to contribute levoglucosan and CO₂. Moreover, the conductive PPy phase provides additional carbon precursors that enhance char formation upon pyrolysis.The third stage (360.4–601.8°C, Δm = 19.4%) represents progressive carbonization and oxidation of the residual framework. In this stage, the PPy-derived carbon undergoes structural reorganization into more condensed aromatic structures, leading to partial graphitization:C(residue) → C(graphitized) + volatile fragmentsThe final char yield is 30.2%, which is significantly higher than the 22% observed for pristine cellulose. This demonstrates the role of PPy in stabilizing the structure against complete volatilization, yielding a more thermally resilient carbon framework.From a kinetic perspective, the Kissinger method gave an Ea of 48.9 kJ·mol⁻¹, which is slightly lower than that of AS (53.9 kJ·mol⁻¹), confirming that the decomposition of PPy requires less activation energy. The Coats–Redfern method produced exceptionally low Ea values (6–9 kJ·mol⁻¹) under diffusion models, reflecting the control exerted by volatile transport in the porous composite. Thermodynamically, the parameters ΔH = 44.2 kJ·mol⁻¹, ΔG = –52.1 kJ·mol⁻¹, and ΔS = +170 J·mol⁻¹·K⁻¹ highlight an endothermic but entropy-driven and spontaneous decomposition process.Comparisons with literature show close agreement: PPy and PPy-based composites typically degrade in the 280–320°C range, release nitrogen-containing volatiles (NH₃, HCN), and yield 25–35% char at 600°C. Our results confirm that the AS/PPy system falls within these ranges while benefiting from the synergistic contribution of cellulose, which provides structural support, and PPy, which improves char yield.Practically, the AS/PPy composite provides a compromise between cellulose and PTSA-doped materials. Its lower Tmax compared to cellulose is offset by its higher char yield, making it suitable for applications where enhanced conductivity and moderate thermal resilience are required, such as electrode materials in supercapacitors, biosensors, and antistatic coatings.Thermal decomposition of AS/PPy–PTSA compositeDoping the PPy phase with p-toluenesulfonic acid (PTSA) introduces ionic moieties into the composite, thereby altering both the onset temperature of decomposition and the pathways by which volatile products are generated (fig.3). The AS/PPy–PTSA system demonstrates a distinctly different profile from both pristine cellulose and the undoped AS/PPy composite.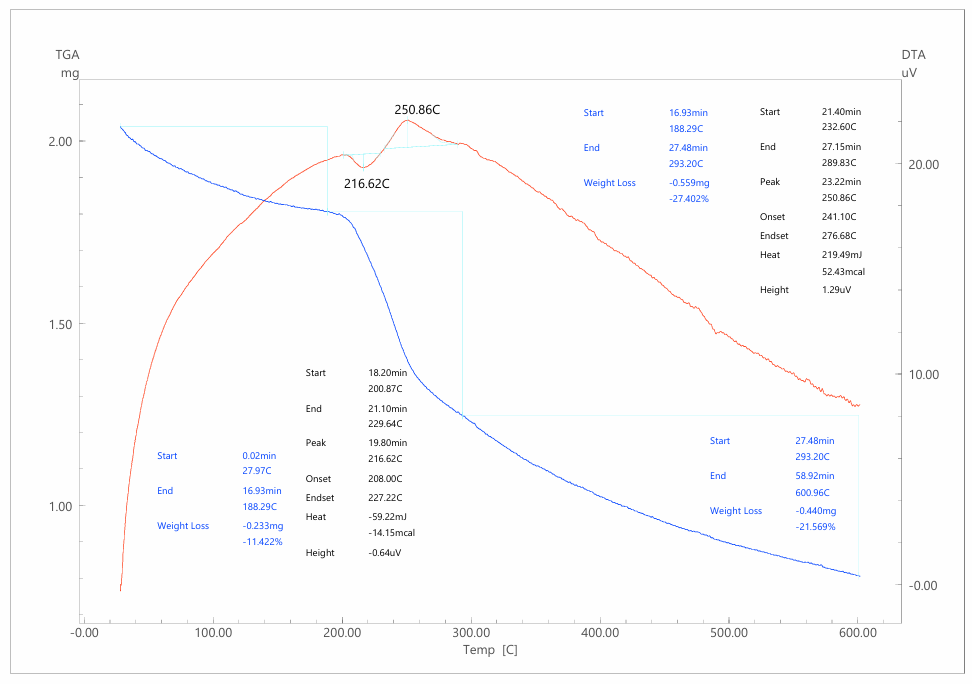 | Figure 3. Derivatogram of the AS/PPy-PTSA composite |
The first stage (27.9–188.3°C, Δm = 11.4%) is strongly influenced by the presence of PTSA. An endothermic peak at 216.6°C reflects the simultaneous dehydration of the composite and partial volatilization/decomposition of free PTSA molecules. The characteristic decomposition of sulfonic acid groups can be represented by the following reaction:CH₃C₆H₄SO₃H → CH₃C₆H₄OH + SO₂ + H₂OThis early onset of mass loss highlights the reduced thermal stability of doped PPy but also introduces sulfur-containing gaseous products (SO₂), which are absent in undoped systems.The second stage (188.3–293.2°C, Δm = 27.4%) is the most intense and is associated with simultaneous degradation of PPy chains and PTSA sulfonic groups. A strong exothermic peak at 250.9°C confirms the highly energetic nature of this process. The mechanism involves chain scission of doped PPy and decomposition of –SO₃H groups:(C₄H₃N)n⁺ [PTS⁻] → PPy fragments + NH₃ + HCN + H₂O–SO₃H → SO₂ + ½ O₂The release of NH₃, HCN, SO₂, and water vapor at this stage accounts for the steep mass loss and strong exothermic response.The third stage (293.2–601.0°C, Δm = 21.6%) corresponds to further carbonization of the residue. Interestingly, the char yield for AS/PPy–PTSA is the highest among the three systems, reaching 39.6%. This suggests that PTSA promotes cross-linking and aromatic condensation during pyrolysis, leading to a more stable carbonaceous network. At elevated temperatures, this residue gradually oxidizes:C(residue) + O₂ → CO₂Kinetic analysis shows that the Kissinger activation energy decreases further to 43.8 kJ·mol⁻¹, indicating that decomposition is easier to initiate compared to both AS and AS/PPy. Coats–Redfern analysis again yields low Ea values (9–13 kJ·mol⁻¹) under diffusion models. Thermodynamic data (ΔH = 39.5 kJ·mol⁻¹, ΔG = –48.3 kJ·mol⁻¹, ΔS = +170 J·mol⁻¹·K⁻¹) confirm an endothermic yet spontaneous process, with a high degree of entropy gain.Comparison with literature data for PTSA-doped conducting polymers, such as polyaniline/PTSA and PPy/PTSA, supports our findings. Previous studies report decreased decomposition temperatures due to the destabilizing effect of ionic doping, coupled with increased char yields owing to enhanced carbonization. Our results align with these trends, demonstrating the universality of the effect.From a practical perspective, the AS/PPy–PTSA composite, despite its lower onset stability, is advantageous in applications where high char yield is beneficial. In supercapacitor electrodes, the formation of stable carbon frameworks ensures good conductivity and capacitance retention after cycling. In EMI shielding, the enhanced char residue provides better barrier properties and fire resistance. The release of sulfur-containing gases during pyrolysis may also find relevance in catalytic or sensing applications.The main parameters of thermal decomposition stages, including temperature intervals, mass losses, and char yields for AS, AS/PPy, and AS/PPy–PTSA, are summarized in Table 1.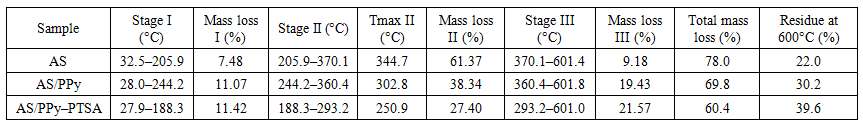 | Table 1. Summary of thermal decomposition stages for AS, AS/PPy and AS/PPy–PTSA (TGA/DTA results) |
Comparative analysisA clear trend emerges across the three systems. The maximum decomposition temperature (Tmax) decreases systematically: 344.7°C (AS) → 302.8°C (AS/PPy) → 250.9°C (AS/PPy–PTSA). At the same time, the char yield increases from 22% for cellulose to 30% for the PPy composite and further to 40% for the PTSA-doped composite. Activation energies follow the same decreasing trend: 53.9 → 48.9 → 43.8 kJ·mol⁻¹.This demonstrates the dual effect of incorporating PPy and PTSA: while reducing the thermal stability at the onset, they promote the formation of a more robust carbonaceous framework at higher temperatures. This trade-off between lower Tmax and higher char yield is of great importance for functional materials.Thermodynamic interpretationAll systems exhibit positive enthalpy (ΔH), negative Gibbs free energy (ΔG), and high positive entropy (ΔS). This combination indicates that the degradation processes require energy input to overcome bond dissociation but are thermodynamically favorable and entropy-driven at elevated temperatures. The nearly identical entropy values (~+170 J·mol⁻¹·K⁻¹) across all samples reflect the commonality of the transition from ordered solid structures to highly disordered gaseous mixtures. The systematic reduction of ΔH and Ea from AS to AS/PPy–PTSA suggests that the presence of conducting polymer chains and sulfonic dopants facilitates the initiation of degradation, while at the same time stabilizing the carbonized residue.The kinetic and thermodynamic parameters calculated from Kissinger and Coats–Redfern analyses are listed in Table 2, confirming the systematic decrease of Ea and ΔH across the studied series. | Table 2. Kinetic and thermodynamic parameters of thermal decomposition |
To better illustrate the observed trends, the comparative indices of thermal stability for the studied systems are presented in Table 3.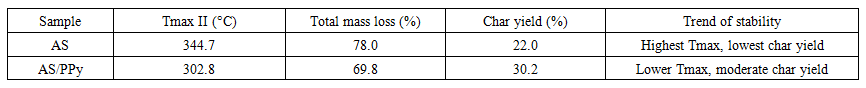 | Table 3. Comparative indices of thermal stability |
Practical implicationsThe practical implications of these findings are highly relevant for advanced applications. For sensors and flexible electronics, where operational temperatures are typically below 150–170°C, the lower Tmax is not critical. Instead, the higher char yield ensures the preservation of electrical conductivity and mechanical integrity under moderate heating. For supercapacitors, the increased carbonization yields stable porous conductive structures after pyrolysis, directly enhancing capacitance and cycling stability. For EMI shielding and protective coatings, the higher char fraction improves fire resistance and long-term durability. Additionally, for catalysis and biomedical applications, the stable carbonized frameworks of PTSA-doped composites offer promising platforms with tailored porosity and chemical functionality.
4. Conclusions
In this study, the thermal behavior of pristine cellulose (AS), cellulose/polypyrrole composite (AS/PPy), and its p-toluenesulfonic acid-doped analogue (AS/PPy–PTSA) was systematically analyzed by TGA/DTA in the temperature range of 25–600°C. All samples exhibited three major decomposition stages, but the introduction of polypyrrole and PTSA significantly altered the degradation pathways, kinetic parameters, and carbonization behavior.Pristine cellulose displayed a major decomposition peak at 344.7°C with a total weight loss of 78% and a low char yield of 22%, confirming its limited thermal resistance. Incorporation of polypyrrole shifted the maximum degradation to 302.8°C, decreased the total weight loss to 70%, and increased the char yield to 30%, indicating the stabilizing role of PPy in promoting carbon residue formation. PTSA doping further reduced the Tmax to 250.9°C but led to the highest char yield of ~40%, demonstrating its dual role in lowering onset stability while enhancing final carbonization.Kinetic and thermodynamic analyses revealed a systematic decrease in activation energy from 53.9 to 43.8 kJ·mol⁻¹ across the series AS → AS/PPy → AS/PPy–PTSA, while ΔH values decreased accordingly. Negative ΔG values confirmed the spontaneous nature of decomposition, and positive ΔS values indicated the entropy-driven character of the process.Overall, the results highlight that although doping reduces initial thermal stability, it significantly enhances residue stability, thereby expanding the functional potential of cellulose-based composites. These findings demonstrate that AS/PPy–PTSA materials are particularly promising for applications in supercapacitors, flexible electronics, sensors, and electromagnetic shielding, where a balance between conductivity, flexibility, and high-temperature resilience is required.
References
| [1] | Poletto, M., Zattera, A. J., Forte, M. M. C., & Santana, R. M. C. (2012). Thermal decomposition of wood: Influence of wood components and cellulose crystallite size. Bioresource Technology, 109, 148–153. https://doi.org/10.1016/j.biortech.2011.11.122. |
| [2] | Yang, H., Yan, R., Chen, H., Lee, D. H., & Zheng, C. (2007). Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel, 86(12–13), 1781–1788. https://doi.org/10.1016/j.fuel.2006.12.013. |
| [3] | MacDiarmid, A. G. (2001). “Synthetic metals”: A novel role for organic polymers. Synthetic Metals, 125(1), 11–22. https://doi.org/10.1016/S0379-6779(01)00507-2. |
| [4] | Huang, W., & Wang, Z. (2011). Polypyrrole–cellulose composites: Synthesis, structure and properties. Carbohydrate Polymers, 84(1), 240–246. https://doi.org/10.1016/j.carbpol.2010.11.045. |
| [5] | Abdelkader, A. H., & El Nashar, R. M. (2014). Conducting polymer composites based on polypyrrole and cellulose for sensing applications. Synthetic Metals, 189, 75–83. https://doi.org/10.1016/j.synthmet.2014.01.010. |
| [6] | Stejskal, J., & Gilbert, R. G. (2002). Polyaniline. Preparation of a conducting polymer (IUPAC Technical Report). Pure and Applied Chemistry, 74(5), 857–867. https://doi.org/10.1351/pac200274050857. |
| [7] | Sapurina, I., & Stejskal, J. (2008). The mechanism of the oxidative polymerization of aniline and the formation of supramolecular polyaniline structures. Polymer International, 57(12), 1295–1325. https://doi.org/10.1002/pi.2484. |
| [8] | Poletto, M., Pistor, V., Zattera, A. J., & Santana, R. M. C. (2011). Thermal decomposition of cellulose in natural fibers. Polímeros, 21(4), 386–391. https://doi.org/10.1590/S0104-14282011005000073. |
| [9] | Kissinger, H. E. (1957). Reaction kinetics in differential thermal analysis. Analytical Chemistry, 29(11), 1702–1706. https://doi.org/10.1021/ac60131a045. |
| [10] | Vyazovkin, S., Burnham, A. K., Criado, J. M., Pérez-Maqueda, L. A., Popescu, C., & Sbirrazzuoli, N. (2011). ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochimica Acta, 520(1–2), 1–19. https://doi.org/10.1016/j.tca.2011.03.034. |

 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML