| [1] | A.A. Spasov, I.N. Yozhitsa, L.I. Bugaeva, V.A. Anisimova, Benzimidazole derivatives: Spectrum of pharmacological activity and toxicological properties, Pharmaceutical chemistry Journal, 33 (1999) 232–243. |
| [2] | R.A. Bucknall, S.B. Carter, A reversible inhibitor of nucleic acid synthsis, Nature, 213 (1967) 1099–1101. |
| [3] | S.O. Podunavac-Kuzmanovic, V.M. Leovac, N.U. Perisic-Janjic, J. Rogan, J. Balaz, Complexes cobalt (II), zinc (II) and copper (II) with some newly synthesized benzimidazole derivatives and their antibacterial activity, Journal of the Serbian Chemical Society, 64 (1999) 381–388. |
| [4] | I.S. Ahuja, I. Prasad, Isonicotinamide complexes with some metal (II) halides and pseudohalides, Inorganic and Nuclear Chemistry Letters, 12 (1976) 777–784. |
| [5] | Ş. Yurdakul, M. Kurt, Vibrational spectroscopic studies of metal (II) halide benzimidazole, Journal of molecular structure, 650 (2003) 181–190. |
| [6] | Lee, Sang Yeon, Bong Hyun Boo, Molecular structure and vibrational spectra of 9-fluorenone density functional theory study. Bull. Korean Chem. Soc, 17(1996) 760–764. |
| [7] | Lee, Sang Yeon, Bong Hyun Boo, Density functional theory study of vibrational spectra of anthracene neutral and radical cati-on, Bull. Korean Chem. Soc, 17.8 (1996) 755. |
| [8] | F.J. Devlin, J.W. Finley, P.J. Stephens, M.J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields: a comparison of local, nonlocal, and hybrid density functionals, The Journal of Physical Chemistry, 99 (1995)16883–16902. |
| [9] | N.C. Handy, C.W. Murray, R.D. Amos, Study of me-thane, acetylene, ethene, and benzene using Kohn-Sham theory, The Journal of Physical Chemistry, 97 (1993) 4392–4396. |
| [10] | N.C. Handy, P.E. Maslen, R.D. Amos, J.S. Andrews, C.W. Murray, G.J. Laming, The harmonic frequencies of ben-zene, Chemical physics letters, 197 (1992) 506–515. |
| [11] | N. Sundaraganesan, S. Ilakiamani, H. Saleem, P.M. Wojciechowski, D. Michalska, FT-Raman and FT-IR spectra, vibration-al assignments and density functional studies of 5-bromo-2-nitropyridine, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 61 (2005) 2995–3001. |
| [12] | M. Goodgame, F.A. Cotton, Preparation and Magnetic and Spectral Studies of Some Cobalt (II) Complexes of Benzimidazole, Journal of the American Chemical Society, 84 (1962) 1543–1548. |
| [13] | M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, J.A. Montgomery, T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar, J. Tomasi, V. Barone, B. Mennu¬cci, M. Cossi, G. Scalmani, N. Rega, G.A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox, H.P. Hratchian, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Strat¬mann, O. Yazyev, A.J. Austin, R. Cammi, C. Po¬melli, J.W. Ochterski, P.Y. Ayala, K. Morokuma, G.A. Voth, P. Salvador, J.J. Dannenberg, V.G. Za¬krzewski, S. Dapprich, A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K. Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J. Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M. Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, C. Gonzalez, J.A. Pople, Gaussian 03, Revision B.04, Gaussian, Inc., Wallingford CT, 2004. |
| [14] | C.T. Lee, W.T. Yang, R.G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev., B 37 (1988) 785–789. |
| [15] | A.D. Becke, Densityfunctional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 98 (1993) 5648–5652. |
| [16] | R.G. Parr, W. Yang, Density-functional theory of atoms and molecules (Vol. 16), Oxford university press, 1989. |
| [17] | P.J. Hay, W.R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg, J. Chem. Phys., 82 (1985) 270–283. |
| [18] | W.R. Wadt, P.J. Hay, Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi, J. Chem. Phys., 82 (1985) 284–298. |
| [19] | P.J. Hay, W.R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 82 (1985) 299–310. |
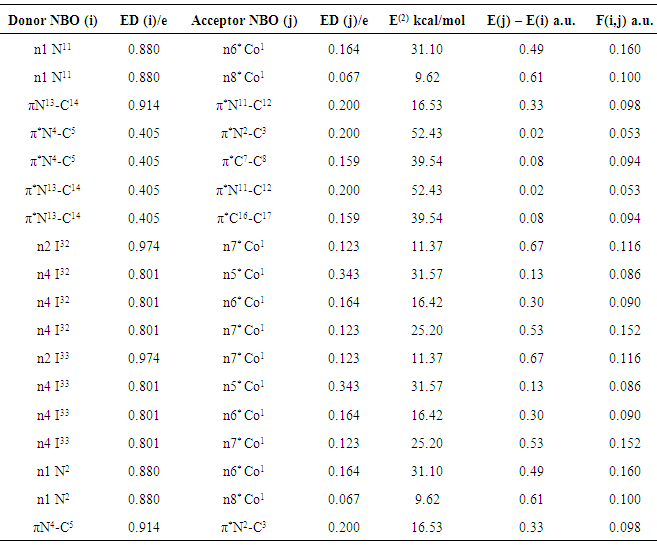
| [20] | A.E. Reed, L.A. Curtiss, F. Weinhold, Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint, Chemical Reviews, 88 (1988) 899–926. |
| [21] | E.D. Gledening, A.E. Reed, J.A. Carpenter, F. Weinhold, NBO. version 3.1. |
| [22] | F. Weinhold, C.R. Landis, Natural bond orbitals and extensions of localized bonding concepts, Chem. Educ. Res. Pract., 2 (2001) 91–104. |
| [23] | M.A. Palafox, J.L. Nunez, M. Gil, Accurate scaling of the vibrational spectra of aniline and several derivatives, Journal of Molecular Structure: THEOCHEM, 593.1 (2002) 101–131. |
| [24] | E. Şahin, S. Ide, M. Kurt, Ş. Yurdakul, Structural investigation of dibromobis (benzimidazole) Zn (II) complex, Journal of molecular structure, 616 (2002) 259–264. |
| [25] | Z.Y. Wu, D.J. Xu, C.H. Hung, Synthesis and crystal structure of diiodobis (thiourea) mercury (ii)-bis (diazafluoren-9-one), Journal of Coordination Chemistry, 57 (2004) 791–796. |
| [26] | C.J. Dik-Edixhoven, H. Schenk, H. Van der Meer, Cryst. Struct. Commun. 2 (1973) 23. |
| [27] | M. Karabacak, M. Çınar, M. Kurt, An experimental and theoretical study of molecular structure and vibrational spectra of 2-chloronicotinic acid by density functional theory and ab initio Hartree–Fock calculations, Journal of Molecular Structure, 885 (2008) 28–35. |
| [28] | Halls, Mathew D., Julia Velkovski, and H. Bernhard Schlegel, Harmonic frequency scaling factors for Hartree-Fock, S-VWN, B-LYP, B3-LYP, B3-PW91 and MP2 with the Sadlej pVTZ electric property basis set, Theoretical Chemistry Accounts, 105.6 (2001) 413–421. |
| [29] | W.R. Augus, C.K. Ingold, A.H. Leekie, J. Chem. Soc, (1936) 925. |
| [30] | C.R. Bailey, S.C. Carson, R.R. Gordon, C.K. Ingold, Structure of benzene. Part XIX. The infrared spectra of 1:4-dideuterobenzene and 1:2:4:5-tetradeuteroben¬zene: description and analysis, Journal of the Chemical Society, 63 (1946) 288–299. |
| [31] | E.F. Mooney, The infrared spectra of chlorobenzene and bromobenzene derivatives–III. Toluenes, Spectrochimica Acta 20.9 (1964)1343–1348. |
| [32] | G. Joshi, N.L. Singh, Infrared absorption spectrum of orthofluorotoluene, Spectrochimica Acta Part A: Molecular Spectroscopy, 23.5 (1967) 1341–1344. |
| [33] | M. Tsuboi, 15 N isotope effects on the vibrational frequencies of aniline and assignments of the frequencies of its nh 2 group, Spectrochimica Acta, 16.4 (1960) 505–512. |
| [34] | C.S. Venkateswaran, N.S. Pandya, The Raman spectra of organic compounds: aniline. Proceedings of the Indian Academy of Sciences-Section A., Springer India, 15(5) (1942) 390–395. |
| [35] | J.C. Evans, The vibrational assignments and configuration of aniline, aniline-NHD and aniline-ND 2, Spectrochimica Acta, 16.4 (1960) 428–442. |
| [36] | S. Pinchas, David Samuel, Marta Weiss-Broday, The infrared absorption of 18 O-labelled benzamide, Journal of the Chemical Society, (1961) 1688–1692. |
| [37] | L. Kahovec, K.F. Kohlrausch, Studien zum Raman-Effekt. Monatshefte für Chemie/Chemical Monthly, 74.1 (1941) 104–117. |
| [38] | V.S. Sastri, J.R. Perumareddi, Molecular orbital theoretical studies of some organic corrosion inhibitors, Corros. Sci., 53 (1997) 617–622. |
| [39] | K.K. Irikura, THERMO.PL, National Institute of Standards and Technology, Gaithersburg, MD, 2002. |
| [40] | J.P. Merrick, D. Moran, L. Radom, An evaluation of harmonic vibrational frequency scale factors, J. Phys. Chem. A, 111 (2007) 11683–11700. |
| [41] | J. Ott, J. Bevan, J. Boerio-Goates, Calculations from Statistical Thermodynamics, Academic Press., 2000. |

 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML