-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Materials and Chemistry
p-ISSN: 2166-5346 e-ISSN: 2166-5354
2015; 5(2): 31-43
doi:10.5923/j.ijmc.20150502.02
Synthesis, Characterization and DFT Calculations of 1,4-diphenyl-3-(p-nitrophenylamino)-1,2,4-triazolium Hydroxide Inner Salt (Nitronitron) by 1H NMR, FT-IR, UV/Vis and HOMO-LUMO Analysis
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLZeyad A. Saleh1, Firyal Weli Askar2, Salah M. A. Ridha3
1Physics Department, College of Science, AL-Mustansiriyah University, Baghdad, Iraq
2Chemistry Department, College of Science, AL-Mustansiriyah University, Baghdad, Iraq
3Physics Department, College of Science, Kirkuk University, Kirkuk, Iraq
Correspondence to: Salah M. A. Ridha, Physics Department, College of Science, Kirkuk University, Kirkuk, Iraq.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Inthis study, the synthesis and characterization of a novel compound, (1,4-Diphenyl-3- (p-nitrophenylamino) -1,2,4-triazolium hydroxide inner salt (Nitonitron), C20H15N5O2) were reported. The spectroscopic investigations of the compound were studied by 1H NMR, FT-IR, UV/Vis techniques. The 1H NMR spectra were recorded in DMSO solution. FT-IR spectrum in solid state was observed in the region 4000-400 cm-1. The UV/Vis absorption spectrum of the compound which dissolved in chloroform was recorded in the range 200-800 nm. The structural data, fundamental vibrational modes, 1H NMR isotropic chemical shifts, frontier orbital energies (HOMO, LUMO), and band gap energy of the compound in the ground state were calculated using density functional theory(DFT) employing B3LYP/6-311G(d,p) basis set. The HOMO and LUMO analysis were used to elucidate information regarding charge transfer within the molecule. Based on the optimized geometry and by using time-dependent density functional theory (TD-DFT) methods in vacuum and solution (chloroform), the allowed excitation and oscillator strengths of electronic absorption spectrum were predicted. The solvation effects have been included by means of the conductor-like polarizable continuum model CPCM. Isotropic chemical shifts were calculated using the Gauge-Independent Atomic Orbital (GIOAO) method. Finally, a comparison between the experimental data and the calculated results appeared a good agreement.
Keywords: Nitronitron, Heterocyclic compound, 1H NMR, IR, UV/Vis, DFT
Cite this paper: Zeyad A. Saleh, Firyal Weli Askar, Salah M. A. Ridha, Synthesis, Characterization and DFT Calculations of 1,4-diphenyl-3-(p-nitrophenylamino)-1,2,4-triazolium Hydroxide Inner Salt (Nitronitron) by 1H NMR, FT-IR, UV/Vis and HOMO-LUMO Analysis, International Journal of Materials and Chemistry, Vol. 5 No. 2, 2015, pp. 31-43. doi: 10.5923/j.ijmc.20150502.02.
Article Outline
1. Introduction
- A large number of heterocyclic compounds, nitrogen-containing heterocycles such as, 1,2,4-triazoles and its derivatives, are an important class of compounds which possess large variety of applications due to their broad-spectrum biological activities, such as antimicrobial [1-3], antibacterial [4, 5], anti-tumor [6], antihypertensive agents [7], antifungal [8-10], anticancer [11], inhibition of vitiligo disease [12], and antiviral [13]. Apart from its biological importance, the use triazole derivatives in the field of anticorrosion protection of metals and alloys [14]. in addition, in 2009, it was demonstrated that triazoles derivatives is used as photosensitizers for dye-sensitized solar cells [15]. To our knowledge, neither the experimental nor computational studies of the synthesized compound of Nitronitron have been available until now.Considering the large physical, biological, and industrial significance, we synthesized Nitronitron based on Nitron by using nitration method. The aim of the present work is to analysis the results obtained from IR, UV/Vis and NMR spectra, and to calculate molecular structure parameters (bond lengths, bond angles, dihedral angles) and fundamental vibrational frequencies associated of Nitronitron compound (Scheme 1).
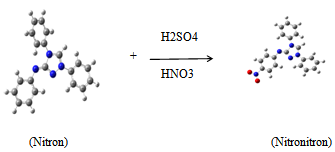 | Scheme 1. Synthesis of Nitronitron |
2. Materials and Methods
2.1. Synthesis
- A mixture of (4.2 ml, 0.087 mole) concentrated sulfuric acid and (9. 6 ml, 0.092mole) nitric acid was added drop by drop of (3.123 gm, 0.01 mole) of Nitron compound with continuous stirring in ice bath for a period of 2 hours, at 5Co. The reaction mixture was poured in to ice, the precipitate was filtered, dried then purified by column chromatography using silica gel eluting with petroleum ether(60-80)°C / benzene (1:1).Yield: 75%.Melting Point: 183-185°C.Color: Yellow FT-IR (KBr, ν, cm-1): 1537 (C=N), 1602(C=C), 1570, 1333(NO2).UV–Vis (Chloroform): λmax /nm (log ε) 430 (0.99). 1H NMR (300 MHz, DMSO-d6, δ, ppm): 8.775 (s, 1H, C1-H), 8.690-8.663 (m,4H), 8.531-7.669 (m, 10H, Ar-H).
3. Experimental Details
3.1. FT-IR Spectrum
- Fourier transform-infrared (FT-IR) measurements were carried out by the KBr method using a Shimadzu corporation 8400S FT-IR spectrometer. FT-IR spectra are generated by the absorption of electromagnetic radiation in the frequency range 4000–400 cm-1.
3.2. UV–Vis Spectrum
- UV/Vis spectrum of the compound has been recorded in the region of 200–800 nm using a Perkin Elmer Lambda 35 UV–Vis spectrometer.
3.3. 1H NMR Spectrum
- 1H NMR spectrum was recorded (299K) on AV300 MHz spectrometer. The compound was dissolved in DMSO. Chemical shifts were reported in ppm relative to tetramethylsilane (TMS) for 1H NMR. 1H NMR spectrum was obtained at a base frequency of 300 MHz.
4. Computational Method
- The molecular geometry, with no symmetry constrains, was optimized using density functional theory (DFT) calculations with a hybrid functional B3LYP (Becke's three parameter exchange functional combined with the LYP gradient corrected correlation functional) at the basis set 6-311G(d.p) by the Berny method [17] to characterized all stationary points as minima. The optimized geometry corresponding to the minimum on the potential energy surface (PES) has been obtained by solving self-consisting field equation iteratively. The calculated vibrational frequencies ascertained that the structure was stable (no imaginary frequency). TD-DFT method is usually found to be a strong and accurate method for describing low-lying excited states of conjugated molecules and has consequently been applied to solve countless chemical and physical problems. Based on the optimized geometry and by using time-dependent density functional theory (TD-DFT) methods in vacuum and solution (chloroform), the allowed excitation and oscillator strengths of electronic absorption spectrum were predicted. The solvation effects have been included by means of the conductor-like polarizable continuum model CPCM [18].In addition, HOMO, LUMO, and energy gap of the title compound were calculated at the B3LYP/6-311G(d,p) level. Finally the Nuclear Magnetic Resonance (NMR) chemical shifts were performed using Gauge Independent Atomic Orbital (GIAO) method [19, 20].
5. Results and Discussion
5.1. Molecular Geometry
- The most optimized structural parameters (bond lengths, bond angles and dihedral angles) of Nitronitron were calculated by DFT/B3LYP level with 6-311G(d,p) basis set and are given in Table 1. The calculated molecular structure of the title compound is found to be non-planar and is as shown in Figure 1. along with the atom numbering scheme. In the literature, we have not found experimental data on molecular structure of Nitronitron, therefore the molecular structure of Nitronitron is compared with the available experimental data of N-(p-dimethylaminobenzylidene)-p-nitroaniline (DBN) and5-[(E)-Methoxy(phenyl)methylidene]-1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazole [21, 22] as crystal data of Nitronitron. As seen in Table 1, all of the bond lengths and bond angles in the phenyl rings are in the normal range. The calculated double N35=C1 and N37=C38 bond lengths were found as 1.3221 and 1.2953 Å, while the single N36-C1, N34-C38 bond lengths in 1,2,4 triazole ring are calculated as 1.3407 and 1.3539 Å, respectively. These single bonds are shorter than expected in triazole ring. This result indicates strong interaction of the filled nitrogen 2p orbital with unoccupied carbon 2p orbital. The calculated single C6-N37 and C3-N40 bond lengths as 1.3778 and 1.4597 Å, respectively. This difference belongs to the fact that electron density is more delocalized on nitro (NO2) group. Also can be seen from the Table 1, in view of the bond length, most of the calculated parameters are longer than experimental ones and the biggest difference occurs at C1-N35, with the different values being 0.078 Å when compared with the experimental values [21, 22]. As for the bond angles by comparing the calculated values with experimental ones, the biggest difference is seen in the bond angle of N(34)- C(38)-N(36), with the difference being 6.19° degree. The (p-nitrophenylamino) fragment at C38 is approximately coplanar with heterocyclic ring. The 1,4 diphenyl rings attached to N35 and N36 subtend dihedral angles of -31.259° and 40.821°, respectively, in relation to the plane of the triazole ring.
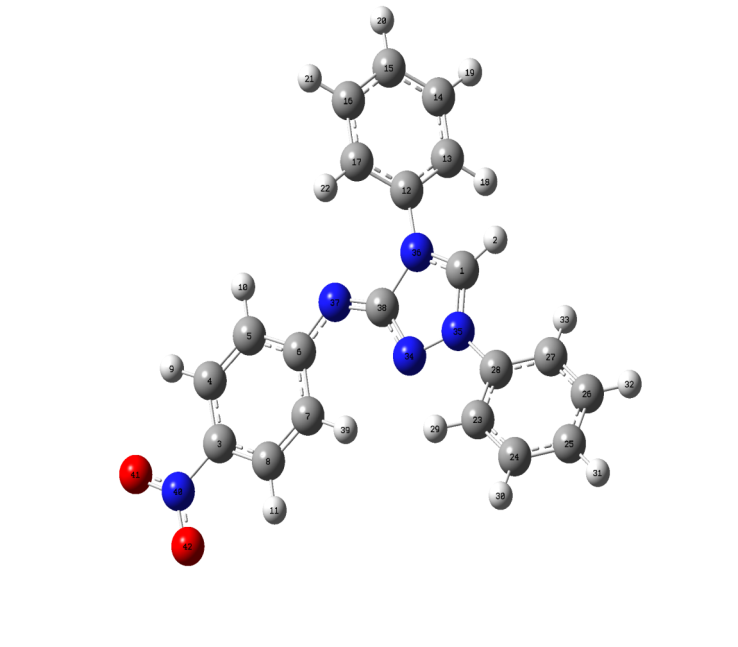 | Figure 1. Molecular structure and numbering scheme of Nitronitron |
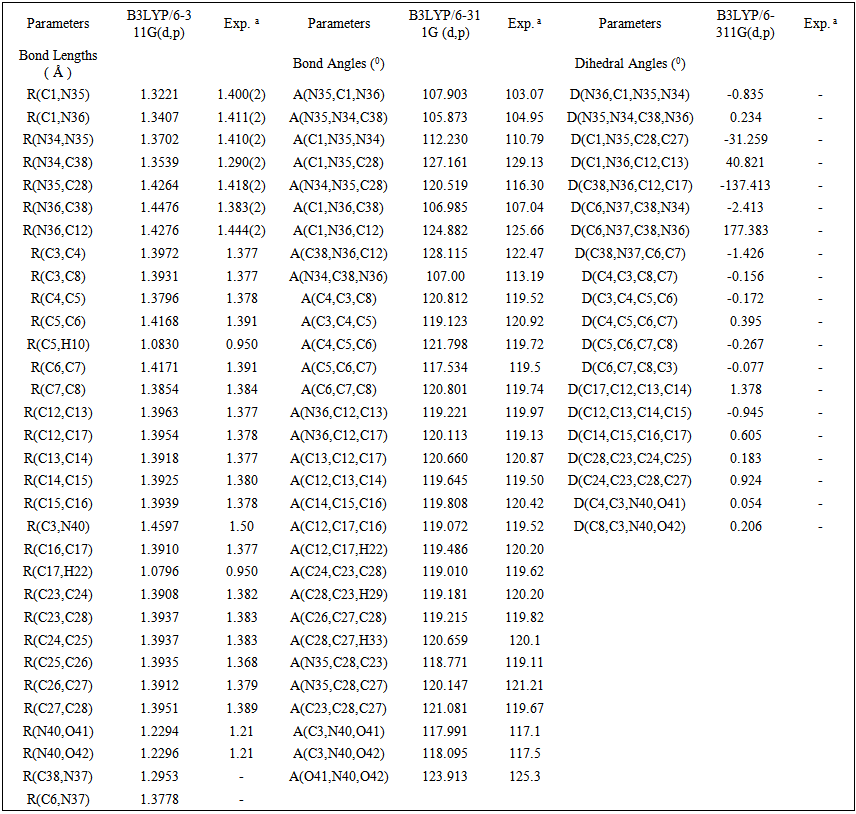 | Table 1. Selected Optimized geometrical parameters of Nitronitron compound in the ground state |
5.2. Vibrational Frequencies
- The Nitronitron molecule have 42 atoms and the number of the vibration modes is 140. All fundamental vibrations which possess C1 symmetry are active in IR spectrum. The observed and calculated vibrational frequencies along with relative intensities, probable assignments at B3LYP/ 6-311G(d,p) level of the title compound are given in Table 2. vibrational frequencies simulated for Nitronitron compound with the unscaled B3LYP/6-311G(d,p) force field are slightly greater than the observed values. These differences can be rectified by scaling the simulated wavenumbers with appropriate factor. It is necessary to scale down the calculated harmonic frequencies to improve the calculated values in agreement with experimental values. The different scale factors can be used for different regions of vibrations to obtain a better agreement between the experimental and computed frequencies. The vibrational frequencies calculated at B3LYP/6-311G(d,p) level were scaled by 0.981 for wave numbers less than 1700 cm-1 and 0.976 for higher wave numbers. Figure 3 shows the experimental and the calculated IR spectrum at B3LYP/6-311G(d,p) level of the title compound.
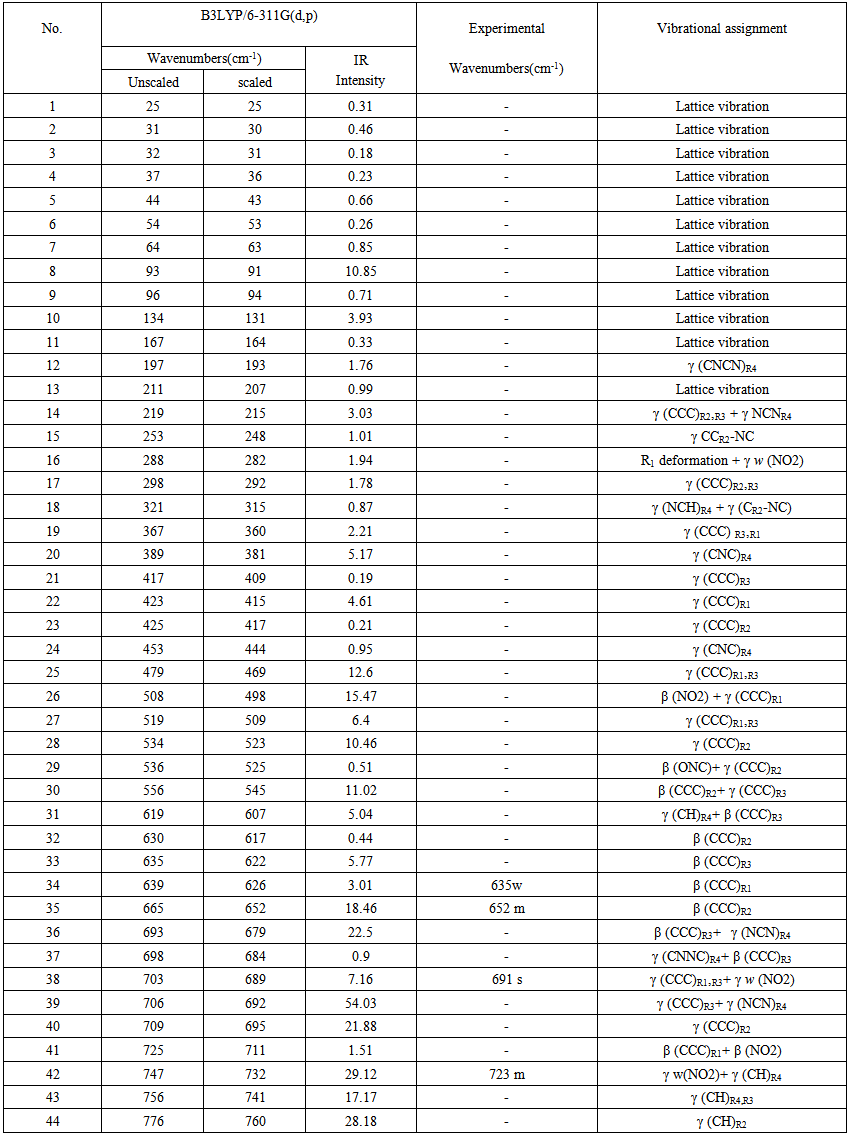 | Table 2. Experimental and calculated IR spectral data of Nitronitron together with their assignment |
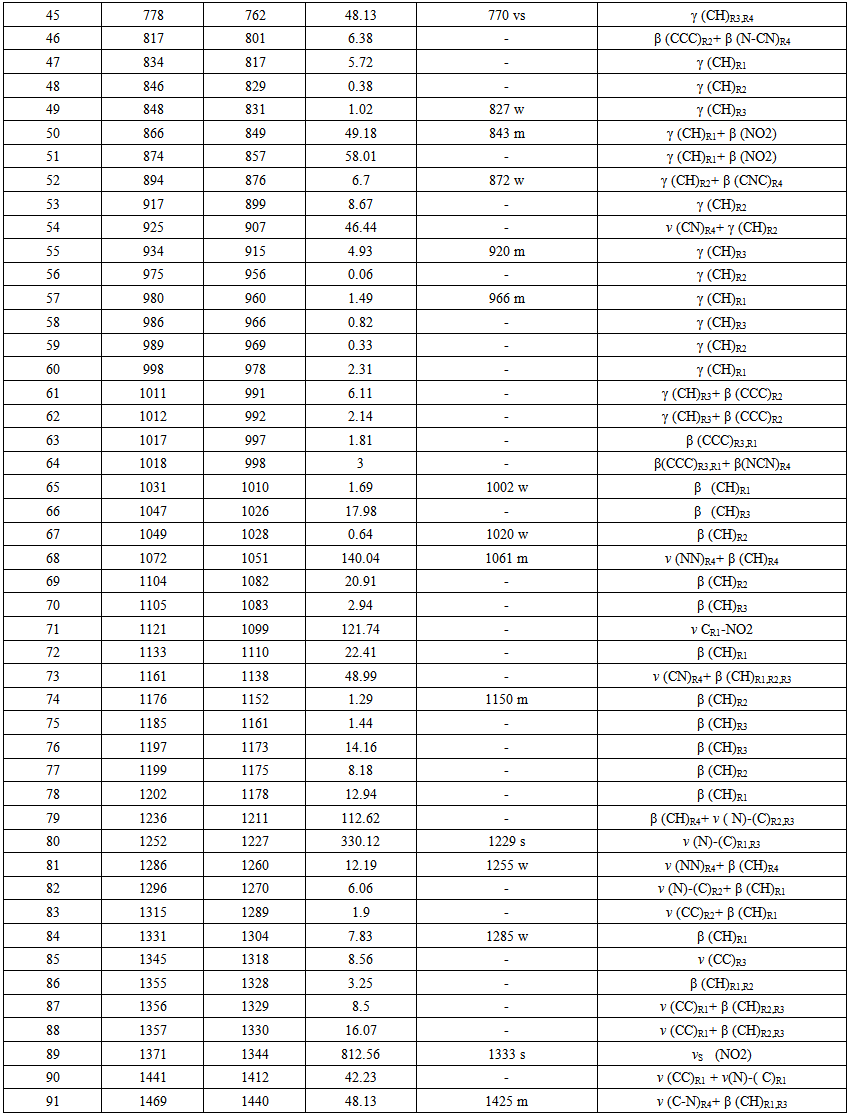 | Table 2. Continued |
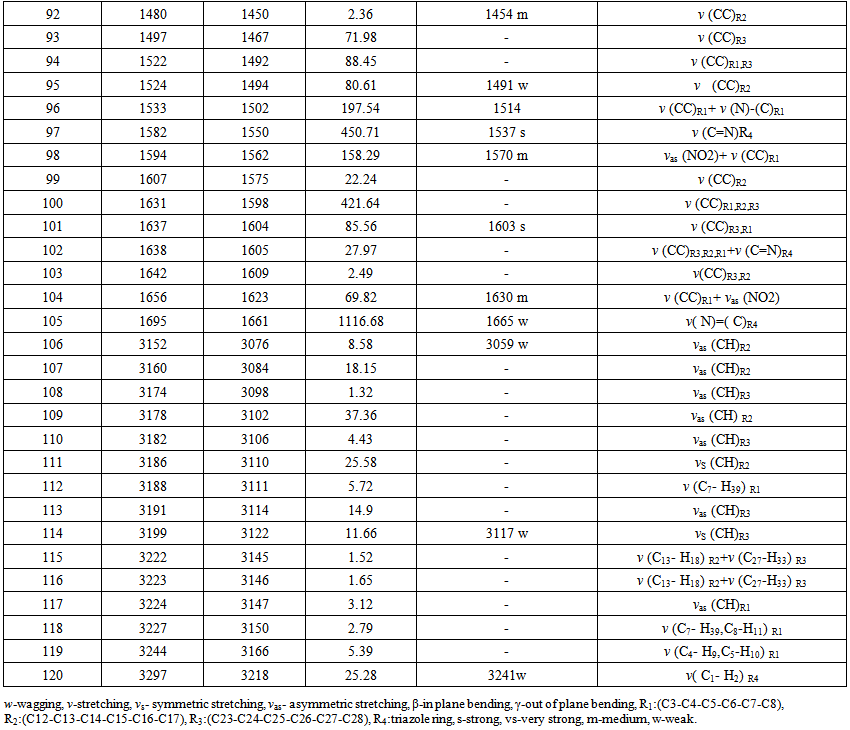 | Table 2. Continued |
5.2.1. C-H Vibrations
- The aromatic compounds shows the appearance of C-H stretching vibrations in the spectral range 3000-3100 cm-1 [23]. The observed bands at 3059, 3117 cm-1 attributed to C-H stretching vibrations of the title compound. DFT computations predict this mode at 3076, 3122 cm-1 for B3LYP/6-311G(d,p) level of theory. The other CH stretching modes (C13-H18, C27-H33, C7-H39, C8-H11, C4-H9, C5-H10, C1-H2) are computed from 3145 to 3218 cm-1. The C-H in-plane bending bands of aromatic compounds are observed in the spectral range 1300-1000 cm-1 [24]. The observed bands at 1002, 1020, 1150, 1285 cm-1 of title compound can attributed to the C-H in-plane bending vibrations. The respective calculated bands are 1010, 1028, 1152, 1304 cm-1 at B3LYP/6-311G(d.p) level for C-H in-plane bending vibrations. The absorption bands arising from C-H out-of-plane bending vibrations are usually observed in the spectral range 1000-750 cm-1 [25]. The computed vibrations at 762, 831, 849, 876, 915, 960 cm-1 are assigned C-H out-of-plane bending vibrations using B3LYP/6-311G(d,p) which are comparable to experimental data at 770, 827, 843, 872, 920, 966 cm-1. The results showed that the theoretical data nearly coincide with experimental ones and these assignments are in good agreement with literature data [24, 25].
5.2.2. C-C Vibrations
- The C=C stretching modes in Benzene rings are in general appear in the spectral range 1650-1400 cm-1[26]. For the title compound the observed C=C stretching vibrations are found at 1454, 1491, 1514 1603, 1630 cm-1 and the respective calculated bands are assigned at 1450, 1494, 1502, 1604, 1623 cm-1. The CCC in-plane bending modes of aromatic rings is observed bands at 635, 652 cm-1 which is comparable to theoretical data at 626, 652 cm-1 and CCCC out-of-plane bending mode is not observed in the spectrum of the title compound. These assignments are in good agreement with B3LYP/6-311G(d,p) method and as well as with the literature data [27].
5.2.3. C-N,C=N, N-N Vibrations
- The recognition of C-N, C=N, and N-N vibrations is a very difficult task, since the mixing of several bands are possible in this region. The observed bands in IR at 1665 cm-1 belong to C(aromatic ring) = N stretching vibration, while the calculated value of this band was appeared at 1661 cm-1 for the title compound. The observed band at 1425 cm-1 can be assigned to the C-N stretching vibration of 1,2,4 triazole ring and the calculated value of the mention mode was found at 1440 cm-1. The C=N stretching mode of triazole ring is observed band at 1537 cm-1, while the calculated value was appeared at 1550 cm-1.The observed band at 1255 cm-1 of title compound can be assigned to the N-N stretching mode and the calculated value of the mentioned mode was appeared at 1260 cm-1. Some of vibrational bands are combined with the other vibrational ones. These assignments are in good agreement with B3LYP/6-311G(d,p) method and as well as with the literature data [28-31].
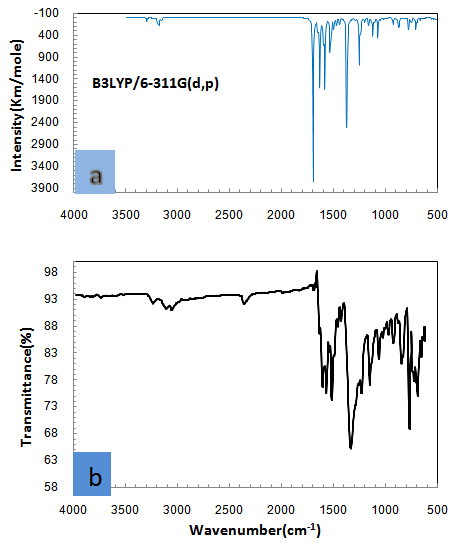 | Figure 2. Theoretical (a) and experimental (b) FT-IR spectra of Nitronitron |
5.2.4. NO2 Group
- Aromatic Nitro(NO2) group compounds show a very strong asymmetric and strong symmetric stretching vibrations at 1540-1614 cm-1 and 1320-1390 cm-1, respectively [32]. The IR spectrum of the Nitronitron two absorption at 1333, 1570 cm-1 with strong and medium have been assigned to NO2 symmetric and asymmetric stretching vibrations, respectively. This resembles the calculated vibrations 1344, 1562 cm-1 obtained from DFT. Nitro (NO2) group scissoring modes is observed at 843, cm-1 combined with other mode and the calculated value of the mention mode was appeared at 849 cm-1. Furthermore, in plane bending (scissors) of nitro group calculated at 711, 857 cm-1. Out of plane bending (wag) modes of nitro group observed at 691 cm-1 combined with the other modes and the calculated value of this mode was appeared at 689 cm-1. Finally, stretching vibration between C(aromatic ring) and nitro group calculated at 1099 cm-1.
5.3. 1H NMR Analysis
- Chemical shifts are recognized as an imperative part of the information contained in NMR spectra. The NMR experimental and theoretical chemical shifts are used to identify the organic compounds. GIAO (Gauge Independent Atomic Orbital) method exhibits a faster convergence of the calculated properties upon extension of the basis set used compared with other methods. GIAO approach is one of the most common methods for calculating isotropic nuclear magnetic shielding tensors. In GIAO method, the atomic basis functions depend explicitly on the magnetic field. 1H NMR chemical shifts of the Nitronitron compound are calculated with GIAO procedure using DFT/B3LYP/6-311G(d,p) method and using Tetramethylsilane (TMS) as reference. The NMR spectra calculations were performed by using Gaussian 09 package. The experimental and theoretical 1H NMR spectra are shown in Figure 3. The calculated 1H NMR showed a good agreement with experimental results obtained for the new synthesized Nitronitron compound named (1,4-Diphenyl-3- (p-nitrophenylamino)-1,2,4-triazolium hydroxide). The obtained theoretical results were helpful for the detailed assignments of experimental 1H NMR spectra of the studied compound. Quantum chemical calculations were used for a better understanding of the NMR properties as well as for an analysis of the geometrical parameters in this novel compound. The H atom is the smallest of all atoms and mostly localized on the periphery of molecules; therefore their chemical shifts would be more susceptible to intermolecular interactions in the aqueous solutions as compared to that for other heavier atoms.
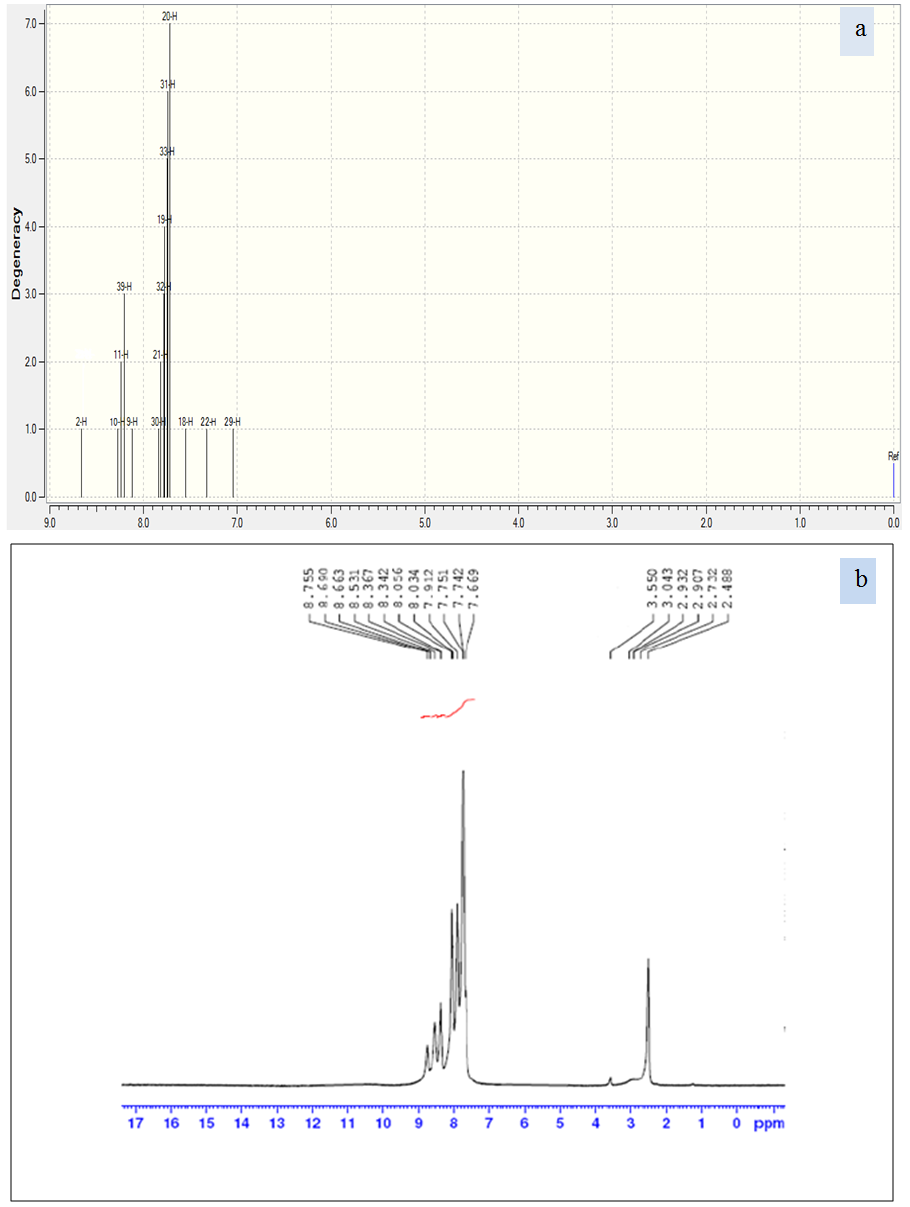 | Figure 3. Theoretical (a) and experimental (b) 1H NMR of synthesized compound in DMSO solvent |
5.4. HOMO-LUMO Analysis
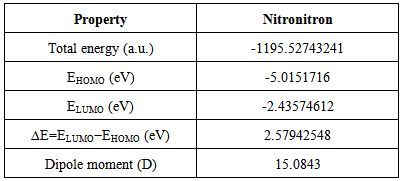
- The frontier molecular orbitals (HOMO and LUMO) are the main orbital participating in chemical reactions and they also used for predicting the most reactive position in π-electron systems. The HOMO energy describes the ability of electron giving, LUMO describes the ability of electron accepting, and the energy gap between HOMO and LUMO describes the molecular chemical stability [34] and it is important parameter in determining molecular electrical transport properties because it is a measure of electron conductivity.In order to investigate the energetic behavior and dipole moment of synthesized compound, optimization were performed. Theoretical calculations included the total energy, HOMO-LUMO energies, energy gap, and dipole moment using B3LYP/6-311G(d,p) level. Results obtained in gas phase are listed in Table 3. and it reveals the energy gap reflect the chemical activity of the molecule. In addition, The energy value of HOMO is computed as (-5.0151716 eV) and LUMO as (-2.43574612 eV), and the energy gap value is (2.57942548 eV) and the dipole moment is (15.0843Debye) in gas phase for the synthesized compound. Lower value in the HOMO and LUMO energy gap explains the eventual charge transfer interactions taking place within the molecule.
|
 -conjugated path.
-conjugated path.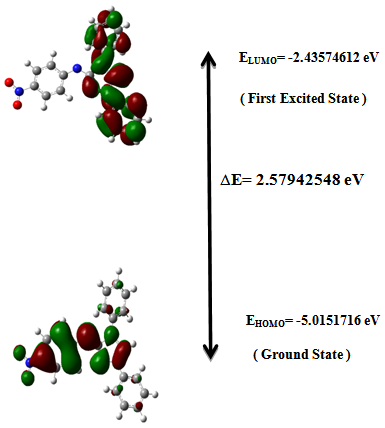 | Figure 4. The atomic orbital components of the frontier molecular orbital of Nitronitron |
5.5. UV/Vis Spectrum
- UV/Vis spectra have been studied using time dependent - density functional theory (TD-DFT)method based on B3LYP/6-311G(d,p) level optimized structure in order to understand electronic transitions of synthesized compound. The experimental UV/Vis spectrum has been observed in chloroform solvent at room temperature, while the theoretical calculations were performed for gas phase and chloroform solvent. The solvent effect was calculated using the conductor-like polarizable continuum model (CPCM). The experimental and theoretical UV/Vis spectra of the synthesized compound is shown in Figure 5. As seen from Figure 5, in experiment, there exist two bands at 340 and 430 nm, respectively. In theory, there is one brad band and the peak is at 374 nm for gas phase, while for solvent case, there was one broad band has red red-shift with value of 419 nm when comparing with gas phase calculations and can easily be seen that solvent case data is the closest with compared to the experimental absorption bands. The error between the calculated and experimental results is 56 nm for gas phase, and 11 nm for solvent case. These values indicate that the solvent case is more suitable than the gas phase for studying the absorption spectra of the title compound. One of the reasons the difference between the experimental values and the theoretical predictions may be TD-DFT calculations do not evaluate the spin-orbit splitting; the values are averaged.
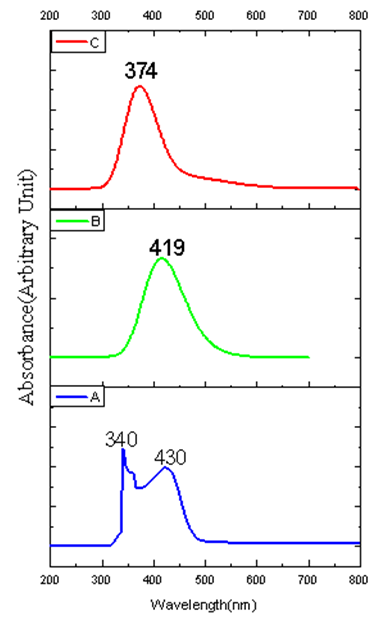 | Figure 5. Experimental and calculated UV/Vis spectra of the synthesized compound: (a) Experimental (chloroform solvent), (b) Theoretical in solvent case (chloroform) and (c) in gas phase |
 transition.
transition.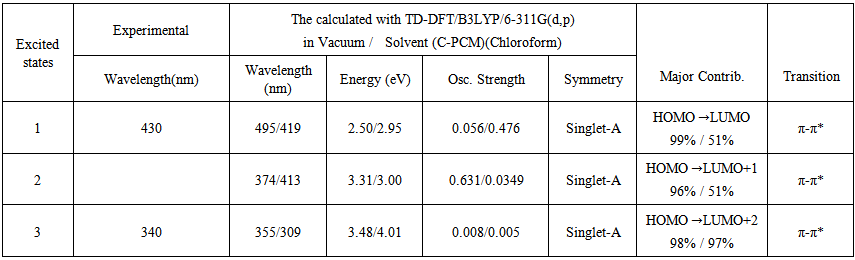 | Table 4. The experimental and calculated absorption wavelength (λ), excitation energies and oscillator strengths (f) of Nitronitron |
4. Conclusions
- The 1,4-Diphenyl-3-(p-nitrophenylamino)-1, 2,4-triazolium hydroxide (Nitronitron) was synthesized and several properties were studied using experimental techniques and computational chemistry calculations for the first time. The geometry was optimized without any symmetry constraints using DFT/B3LYP method with 6-311G(d,p) basis set. The skeleton of the optimized compound is non-planar. On the same base of optimized geometry, 1H NMR were calculated by using the gauge independent atomic orbital (GIAO)/solvent (DMSO) method and their spectra were simulated and the chemical shifts related to TMS were compared with experimental data, showing an acceptable agreement. The vibrational FT-IR spectrum of compound was recorded and assigned with help of the experimental and computed vibrational wavenumbers. The UV/Vis absorption spectrum was examined in chloroform solvent and compared with the calculated one in gas phase and solvent case using TD-DFT/CPCM approach. The experimental band at 430 nm is attributed mainly to a HOMO-LUMO transition is predicted as π-π* transition. The comparison of the predicted bands was done and shows a good agreement. The calculated HOMO and LUMO energies and energy gap analysis show that the occurring of charge transformation within the molecule. Besides the energies of HOMO and LUMO are negative, which indicate that the synthesized compound is stable.