-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Materials and Chemistry
p-ISSN: 2166-5346 e-ISSN: 2166-5354
2015; 5(2): 25-30
doi:10.5923/j.ijmc.20150502.01
Density Functional Theory Calculations of [Me(3-Amino-1,2,4-Triazole)2]2+ Complex Ions (Me =Zn,Cu, Co, Ni and Cd) in Water Phase
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLRumyana Yankova 1, Lachezar Radev 2
1Department of Inorganic and Analytical Chemistry, Assen Zlatarov University, Bourgas, Bulgaria
2Department of Fundamental Chemical Technology, University of Chemical Technology and Metallurgy, Sofia, Bulgaria
Correspondence to: Rumyana Yankova , Department of Inorganic and Analytical Chemistry, Assen Zlatarov University, Bourgas, Bulgaria.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
The quantum chemical calculations of 3-Amino-1,2,4-triazole were made by Hartree-Fock (HF) and Density Functional Theories (DFT) at the B3LYP level with 6-31G(d,p) basis set. From the calculated electrostatic potential and the net atomic charges of 3-Amino-1,2,4-triazole was found that the site most suitable for creation of a coordination bond is N4. The geometric optimization of [Me(3-Amino-1,2,4-triazole)2]2+ complex ions (Me = Zn, Cu, Co, Ni, and Cd) in water phase was done by DFT using Becke’s three-parameter hybrid functional with 6-31G(d,p)basis set and LANL2DZ effecitive core potential for the metals - Co, Cd, and Ni. The bond orders and the electronic properties of the complex ions were calculated. The relationship between the stability constants of the complexes and the electronic properties of the complex ions were examined. It was found that the stability constants of the complexes correlate well with the calculated bond orders Me–L.
Keywords: [Me(3-Amino-1,2,4-triazole)2]2+ complex ions, Quantum chemical calculations, Geometry optimization, Electronic properties
Cite this paper: Rumyana Yankova , Lachezar Radev , Density Functional Theory Calculations of [Me(3-Amino-1,2,4-Triazole)2]2+ Complex Ions (Me =Zn,Cu, Co, Ni and Cd) in Water Phase, International Journal of Materials and Chemistry, Vol. 5 No. 2, 2015, pp. 25-30. doi: 10.5923/j.ijmc.20150502.01.
1. Introduction
- The 5-membered nitrogen heterocyclic rings are structural fragments in a number of biologically active compounds [1], pesticides [2], pigments and other substances used in industry [3, 4].One of the interesting pesticides is 3-Amino-1,2,4-triazole. Probably the most dramatic effect of amino-triazole on growing plants is its interference with chlorophyll formation. This property has simulated the investigation of the compound as a herbicide, cotton defoliant, and inhibitor of second growth. Triazole is a promising substance for commercial control of perennials such as Canada thistle [5], quack-grass [6], Johnson grass [7], nut grass [8], and woody plants like poison ivy and poison oak [9].The pesticides contained in soil often react with metal ions. The information about the geometry and stability constants of such complexes would provide a possibility to describe pesticides propagation in soil and subterranean water, as well as the mechanisms of their transportation in plants.In a previous work, we have studied amino-triazole complex formation in aqueous solutions with ions contained in soil as macro-components (Fe3+, Al3+, Ca2+) and micro-components (Cu2+, Co2+, Cd2+, Ni2+, Zn2+ and Hg2+) [10].The aims of the presented study are to determine the geometric and electronic structure of [Me(3-Amino-1,2,4-triazole)2]2+ complex ions (Me =Zn, Cu, Co, Ni and Cd) in water phase and to establish the relationship between the stability constants of the complexes and the electronic properties of the complex ions by quantum chemical calculations.
2. Computational Methods
- The full optimization of 3-Amino-1,2,4-triazole was carried out by Hartree-Fock (HF) and Density Functional Theories (DFT) methods, using Gaussian 03 software [11] at the B3LYP level with 6-31G(d,p) basis set. All calculations were converged to 10–8 a.u. Vibration frequencies were also calculated to the structure with optimized geometry and no imaginary frequency were obtained, so the stationary point correspond to the minima of the potential energy surface. An effective method for studying the reaction behavior of molecules is the measuring of their electrostatic potential. The electrostatic potential of 3-Amino-1,2,4-triazole was calculated by DFT method at the B3LYP level with 6-31G(d,p) basis sets. The electrostatic potential is widely used in studies on biological systems to predict the reactivity of numerous chemical systems in electro- and nucleophilic reactions [12]. In order to characterize the electronic population on each atomic centre a Mulliken population analysis [13] was carried out for 3-Amino-1,2,4-triazole. The geometric optimization of [Me(3-Amino-1,2,4-triazole)2]2+ complex ions (Me = Zn, Cu, Co, Ni, and Cd) in water phase was done by DFT using Becke’s three-parameter hybrid functional with 6-31G(d,p)basis set and LANL2DZ effecitive core potential for the metals - Co, Cd, and Ni. To take into account the effect of the solvent by self-consistent reaction field (SCRF) the method of Onsager was used [14]. The Onsager model places the solute in a spherical cavity within the solvent reaction field. The solvent is treated as polarizable continuum with a dielectric constant - , instead of explicit solvent molecules. The charge distribution of the solute polarizes the solvent producing a reaction potential. The reaction potential of solvent alters the solute. This interaction is represented by Hamiltonian containing the solvent reaction potential. All calculations are for complexes at ratio Me: L = 1:2.The software packages HyperChem 5.0 [15] and Molekel 5.4 [16] were used for data preparation and visualization of the results.
3. Results and Discussion
- The visualization of the optimized geometrical structure and atomic labeling of 3-Amino-1,2,4-triazole are presented in Figure 1.To choose suitable calculation method, geometric optimization of 3-Amino-1,2,4-triazole carried out and the results were compared with theexperimental results obtained as it is given in Table 1.
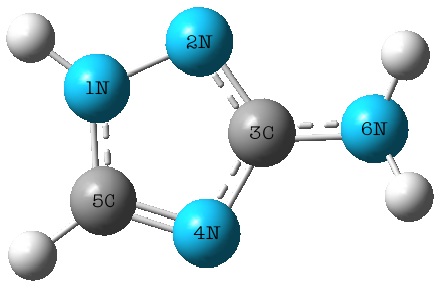 | Figure 1. Optimized geometrical structure and atomic labeling of 3-Amino-1,2,4-triazole |
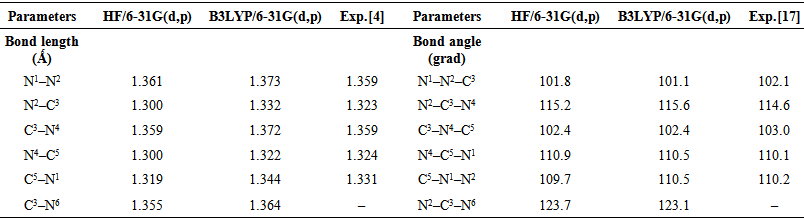 | Table 1. Optimized geometrical parameters of 3-Amino-1,2,4-triazole |
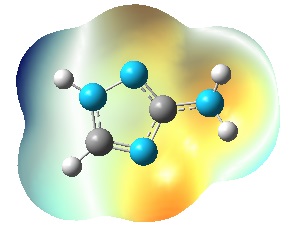 | Figure 2. Electrostatic potential on the surface of3-Amino-1,2,4-triazole |
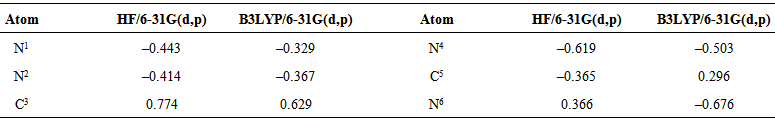 | Table 2. Mulliken atomic charges of 3-Amino-1,2,4-triazole |
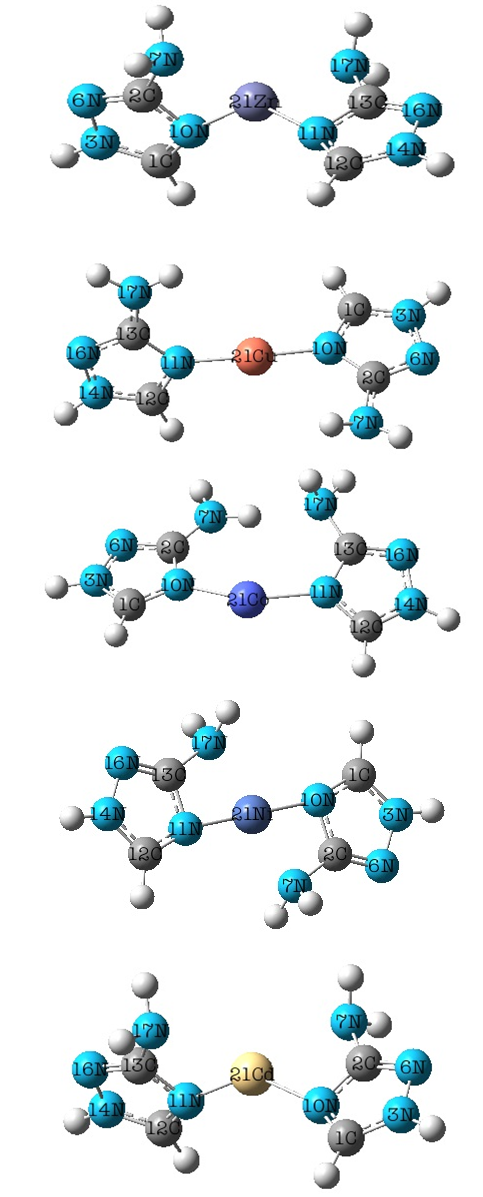 | Figure 3. Optimized geometrical structures and atomic labeling of [Me(3-Amino-1,2,4-triazole)2]2+ complex ionsin water phase |
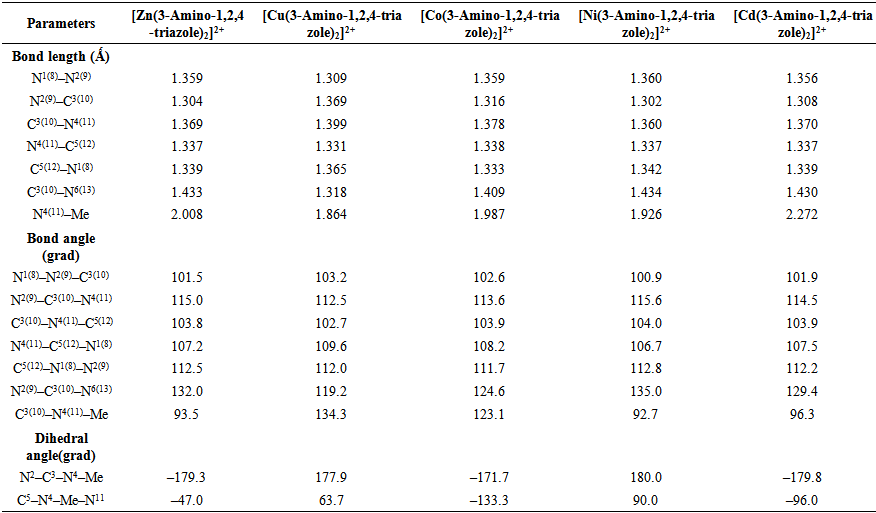 | Table 3. Optimized geometrical parameters of [Me(3-Amino-1,2,4-triazole)2]2+complex ions in water phase |
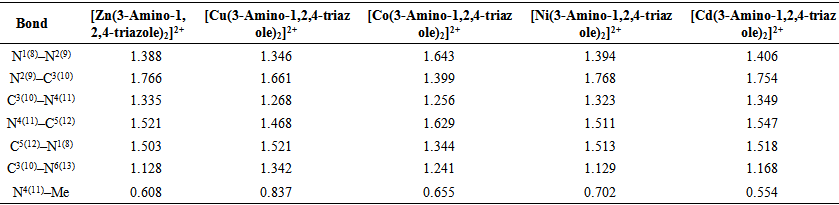 | Table 4. Bond orders of [Me(3-Amino-1,2,4-triazole)2]2+complex ions in water phase |
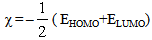 | (1) |
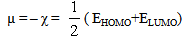 | (2) |
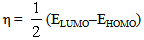 | (3) |
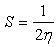 | (4) |
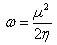 | (5) |
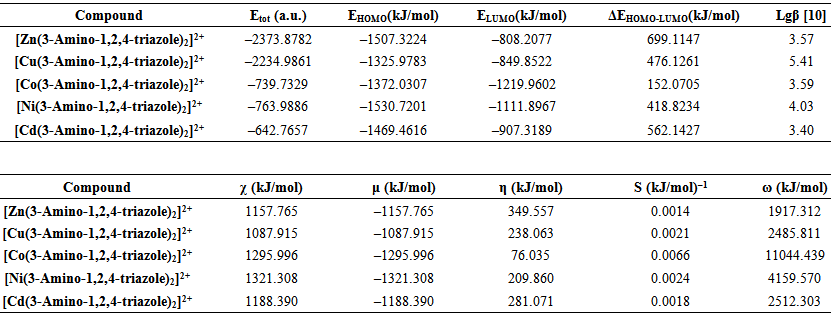 | Table 5. Calculated electronic properties of [Me(3-Amino-1,2,4-triazole)2]2+complex ions in water phase |
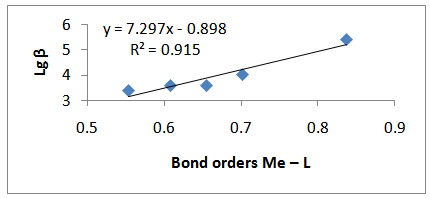 | Figure 4. Relationship between the stability constants of the complexes and the calculated bond ordersMe– L |
4. Conclusions
- The pesticides contained in soil often react with metal ions. The information about the geometry and stability constants of such complexes would provide a possibility to describe pesticides propagation in soil and subterranean water, as well as the mechanisms of their transportation in plants. The geometric optimization of 3-Amino-1,2,4-triazole was done by Hartree-Fock (HF) and Density Functional Theories (DFT) at the B3LYP level with 6-31G(d,p) basis set. From the calculated electrostatic potential and the net atomic charges of 3-Amino-1,2,4-triazole was found that the site most suitable for creation of a coordination bond is N4. The geometric optimization of [Me(3-Amino-1,2,4-triazole)2]2+ complex ions (Me = Zn, Cu, Co, Ni, and Cd) was done in water phase by DFT using Becke’s three-parameter hybrid functional with 6-31G(d,p) basis set and LANL2DZ effecitive core potential fot the metals - Co, Cd, and Ni. The bond orders and the electronic properties of the complex ions were calculated. The relationship between the stability constants of the complexes and the electronic properties of the complex ions were examined. It was found that the stability constants of the complexes correlate well with the calculated bond orders Me–L.