-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Materials and Chemistry
p-ISSN: 2166-5346 e-ISSN: 2166-5354
2012; 2(1): 1-9
doi: 10.5923/j.ijmc.20120201.01
In VitroBioactivity of Collagen/Calcium Phosphate Silicate Composites, Cross-Linked with Chondroitin Sulfate
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLLachezar Radev 1, Nasser Y. Mostafa 2, Irena Michailova 1, Isabel M. M. Salvado 3, Maria H. V. Fernandes 3
1Department of Fundamental Chemical Technology, University of Chemical Technology and Metallurgy, Sofia, 1756, Bulgaria
2ChemistryDepartment, Faculty of Science, Taif University, Taif 888, Kingdom of Saudi Arabia
3Department of Silicate Technology, University of Aveiro and CICECO, Aveiro, 3810-193, Portugal
Correspondence to: Lachezar Radev , Department of Fundamental Chemical Technology, University of Chemical Technology and Metallurgy, Sofia, 1756, Bulgaria.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
In this work we present the experimental results on synthesis structure and in vitro bioactivity of collagen (C)/calcium phosphate silicate (CPS)/chondroitin sulfate (ChS) composites with C/CPS ratio of 25:75 wt. % and 75:25wt. %. The synthesized composites were characterized by XRD, FTIR and SEM/EDS. XRD and FTIR before in vitro test proved that the carbonate containing hydroxyapatite (CO3HA) formed in situ. After in vitro test for 3 days in 1.5 SBF, the nucleation of B-type CO3HA was estimated on the obtained C/CPS=75:25 wt. % composite with cauliflower-like assembly. On the C/CPS=25:75 wt. % amorphous hydroxyapatite (HA) layer was observed.
Keywords: Collagen, Calcium Phosphate Silicate, in Vitro Bioactivity, Chs
Article Outline
1. Introduction
- It should be remembered that proteoglycans have been implicated in the process of regulation of hydroxyapatite (HA) formation and growth in the process of cartilage calcification. In series of papers S.-H. Rhee and J. Tanaka examined the inhibitory effect of chondroitin sulfate (ChS) on HA growth via regulation by the functional groups (COO- and –SO3-) of ChS template[1-3]. On the base of FTIR results they proved that the COO- and –SO3- ions showed the “red shift”, i. e. formed the chemical interactions between HA crystals and them during the precipitation process[1-3]. H. Jiang et al. studied the nucleation kinetics of HA and quantified the effect of ChS on HA nucleation[4]. The experimental results obtained demonstrated that ChS can suppress the supersaturation-driven mismatch and promote of a highly ordered HA crystalline assembly[4]. X. Xiao et al. also demonstrated that the concentration of ChS significantly affects the morphology of produced HA/ChS composites[5]. They also proved that the prepared HA in the presence of ChS is carbonate containing hydroxyapatite (CO3HA)[5]. Others investigated the crystallization behavior of HA/ Collagen (HA/Coll) gels in the presence of ChS under physiological conditions. They concluded that ChSplay an important role in the regulation of biomineralization[6]. S–H. Rhee et al. prepared 80HA/8Coll/12ChS (wt. %) nanocomposite via co-precipitation method from Ca(OH)2, H3PO4 solution containing collagen and ChS, respectively[7]. On the base of the obtained results they proved that when short ChS fibres are mixed with long collagen fibres, the short ChS fibres might be bound to the long collagen fibres with the same axis direction. After that HA crystals start to growth on the collagen fibres[7]. M. Chang modified gelatin (Gel) matrix by the introduction of ChS and then he studied the formation of HA/Gel/ChSnanocomposite[8]. He prepared this composite by mixing of Ca(OH)2 with aqueous solution of H3PO4, gelatin and ChS, respectively and proved that the incorporation of ChS into the Gel matrix contributed to the increase of the organic/inorganic interaction between HA and organic matrix, composed by Gel and ChS[8].In our previous works we have synthesized some bioactive calcium phosphate silicate (CPS) ceramics[9-11]. We observed that the prepared ceramics were in vitro bioactive for different periods of time. On this base we have prepared in vitro bioactive composites with some biodegradable polymers without[12-15] and with[16] cross-linkage.The purpose of our article is to prepare, characterize and evaluate the in vitro bioactivity of collagen/calcium phosphate silicate (C/CPS) composites in the presence of ChS.
2. Experimental Part
- Preparation of the compositesThe CPS ceramic powder as an inorganic part of the composites obtained has been synthesized via polystep sol-gel method with Ca/P+Si molar ratio 1.89. The composition of the gel obtained was 62.6 wt. % CaO, 26.8 wt. % SiO2 and 10.6 wt. % P2O5. The procedure for the synthesis and evaluation of the structure of the prepared CPS ceramics was described in our previous work[9]. Collagen type I, taken in amounts corresponding to the composite content was diluted in 15 ml CH3COOH for 24 h at room temperature under intensively stirring. The C/CPS composite samples, in two types of proportions 25:75 wt. % and 75:25 wt. % were synthesized by adding CPS powder to the C solution with constant stirring for 6 h. pH of the obtained mixture was equal to 2. After homogenation time, pH was adjusted to 9 using 25% NH4OH. ChS was added as cross-linking agent in the quantity equal to 5% degree of linkage, calculated on the base of terminated NH2 groups of collagen. The composite materials obtained were dried at 70oC for 12 h under vacuum.In vitro test for bioactivityBioactivity of the composites obtained was evaluated by examining the apatite formation on their surfaces in 1.5 SBF solutions. The 1.5 SBF was prepared from different reagents as follow: NaCl = 11.9925 g, NaHCO3 = 0.5295 g, KCl = 0.3360 g, K2HPO4.3H2O = 0.3420 g, MgCl2.6H2O = 0.4575 g, CaCl2.2H2O = 0.5520 g, Na2SO4=0.1065 g, and buffering at pH 7.4 at 36.5℃ with 9.0075 g of tris (hydroxymethyl) aminomethane (TRIS) and 1M HCl in distilled water. A few drops of 0.5% NaN3was added to the 1.5SBF solution to inhibit the growth of bacteria[17].The synthesized composites were pressed at 50 MPa with PVA to obtain disc specimens (12 mm diameter and 2 mm thick) and then immersed in 1.5SBF at the human body temperature (36.6℃) in polyethylene bottles in a static condition for 3 days. After soaking the specimens were removed from the fluid, gently rinsed with distilled water, and then dried at 36.6℃ for 12h.Methods for analysisThe structure and in vitro bioactivity of composite materials obtained was monitored by X-ray diffraction (XRD) analysis, Fourier transform infrared (FTIR) spectroscopy and scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDS). Powder XRD spectra were collected within the range from 10 to 80° (2θ) with a constant step 0.04° (2θ) and counting time 1s/step on a Bruker D8 Advance diffractometer with CuKα radiation and SolX detector. The spectra were evaluated with the Diffracplus EVA package. FTIR transmission spectra for the samples obtained were recordered by using of aBruker Tensor 27 spectrometer with a scanner velocity 10kHz. KBr pellets were prepared by mixing ~1 mg of the samples with 300 mg KBr. Transmission spectra were recordered using MCT detector with 64 scans and 1 cm-1 resolution. SEM technique was employed to observe the particle-size and agglomeration of the synthesized composites before and after in vitro test in 1.5 SBF solution. For this a very small amount of a powder was placed on an adhesive carbon tape, coated with gold for 1 min and then observed in a JEOL SEM (model JSM-35 CF, Japan). Energy dispersive X-ray spectroscopy (EDS) was performed for the chemical microanalysis using EDAX system attached to the SEM. After 3 days of immersion, the samples were taken out from 1.5 SBF and the ion concentrations of Ca, P and Si in the solutions were measured by inductively coupled plasma atomic emission spectroscopy (ICP-AES, IRIS, 1000, Thermo Elemental, USA).
3. Results and Discussionanalysis of the Composites before in Vitro Test
- XRD data for the prepared C/CPS samples with different quantity of the organic/inorganic components are given in Fig. 1
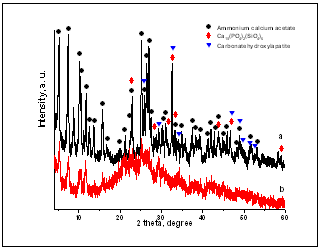 | Figure 1. XRD for the C/CPS samples with 25:75 wt. % (a) and 75:25 wt. % (b) |
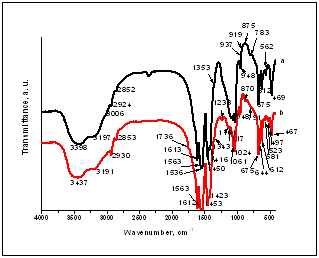 | Figure 2. FTIR spectra for the C/CPS composites: 25:75 wt. % (a) and 75:25 wt. % (b) |
 | Figure 3. SEM of the C/CPS=25:75 wt. % at 500x (a) and 10 000x (b) |
 | Figure 4. SEM of the C/CPS=75:25 wt. % at 5000x (a) and 5000x (b) |
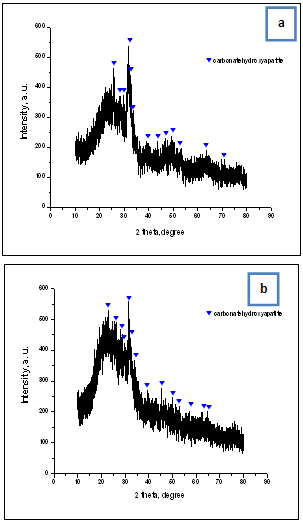 | Figure 5. XRD for the C/CPS=25:75 wt. % (a) and C/CPS=75:25 wt. % (b) after soaking in 1.5 SBF for 3 gays |
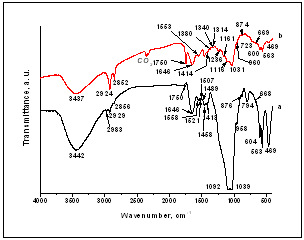 | Figure 6. FTIR spectra of the C/CPS=25:75 wt. % (a) and C/CPS=75:25 (b) after in vitro test in 1.5 SBF for 3 days |
 | Figure 7. SEM of the C/CPS=75:25 wt. % at 5000x (a), 20 000x (b) and for C/CPS=25:75 wt. % at 10 000x (c) after in vitro test in 1.5 SBF for 3 days in a static condition |
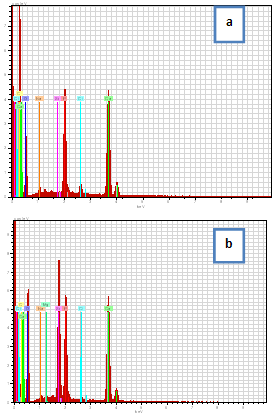 | Figure 8. EDS spectra of the C/CPS=75:25 wt. % (a) and C/CPS=25:75 wt. % (b) |
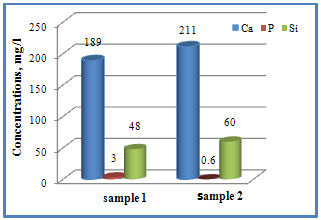 | Fugure 9. Changes in Ca, P and Si contents from C/CPS=75:25wt. % (sample 1) and C/CPS=25:75 wt. % (sample 2) |
4. Conclusions
- The main purpose of the presented work was to synthesize, characterize and assess the in vitro bioactivity of the composites between C and CPS in the presence of ChS. Collagen type I was diluted in 5M CH3COOH and mixed with CPS powder in a 25:75 and 75:25 wt. % ratio in the presence of ChS. XRD for the samples before in vitro test proved that three main phases ammonium calcium acetate, Ca15(PO4)2(SiO4)6 and CO3HA were observed. FTIR depicted that the chemical interaction between COO- from ChS and Ca2+ from partially dissolved CPS ceramic is proved. β-sheet structures in the collagen might also occur. On the same time Ca-def. HA is formed in situ under preparation condition. SEM depicts of the presence of the particles with different morphology. In the C/CPS=75:25 wt. % sample big spherical particles with a bright colour are present, but in the C/CPS=25:75 wt. % some spherical particles are formed on C fibrils. The observation confirmed XRD and FTIR results.After in vitro test in 1.5 SBF for 3 days XRD proved well distinguished HA crystalline phases. On the base of these results we are going closer to the conclusion that the synthesized C/CPS/ChS composites are in vitro bioactive. FTIR analysis determined that the observed “blue shift” for amide I could be assigned to the HA formation on the C/CPS samples. SEM depicts that two kinds of surface particles with different morphology are depicted – big aggregates with cauliflower-like assembly and fully covered surface with quasi-spherical particles. EDS proved the Ca/P ratios 1.67 and 1.33, respectively. On the base of the EDS results we can assume that HA and ACP layers were covered the surface of the synthesized composites. ICP-AES results confirmed XRD, FTIR and SEM results and leads us to conclude that Si-CO3HA may be formed in the immersed C/CPS composites in the presence of ChS.