-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Internal Medicine
p-ISSN: 2326-1064 e-ISSN: 2326-1072
2020; 9(1): 20-22
doi:10.5923/j.ijim.20200901.03

Sharp Syndrome: About a Diagnostic Case at the Medical- Surgical Clinic of the Sylvanus Olympio University Hospital in Lome
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLEdem Komi Mossi1, 2, Salahoudine Maman2, Abdou Razak Moukaïla2, Sodjehoun Apeti2, Dzidzonou Komi Nemi2, Laune Odilon Blatome2, Agbeko Kodjo Djagadou2, Eric Gumedzoe1, Abago Balaka2, Awalou Mohaman Djibril1, 2
1Medical- Surgical Clinic, University Hospital Sylvanus Olympio of Lome, Lome, Togo
2Department of Internal Medicine, University Hospital Sylvanus Olympio of Lome, University of Lome, Lome, Togo
Correspondence to: Edem Komi Mossi, Medical- Surgical Clinic, University Hospital Sylvanus Olympio of Lome, Lome, Togo.
| Email: |  |
Copyright © 2020 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Sharp syndrome is the association in a patient of symptoms identical to at least two connective tissue diseases. It is rare in black Africans. It is characterized by a clinical polymorphism that is often the cause of diagnostic delay. Sharp syndrome is associated with the presence in the blood of antinuclear antibodies with speckled fluorescence whose antigen corresponds to a ribonucleoprotein. The diagnosis is based on the use of diagnostic criteria. We report a case of Sharp syndrome in a 39-year-old patient, whose diagnosis was raised on the basis of diagnostic criteria of Alarcon-Segovia and confirmed by those of the Japanese Ministry of Health and Welfare.
Keywords: Sharp, Polyarthritis, Deterioration of the general condition, Togo
Cite this paper: Edem Komi Mossi, Salahoudine Maman, Abdou Razak Moukaïla, Sodjehoun Apeti, Dzidzonou Komi Nemi, Laune Odilon Blatome, Agbeko Kodjo Djagadou, Eric Gumedzoe, Abago Balaka, Awalou Mohaman Djibril, Sharp Syndrome: About a Diagnostic Case at the Medical- Surgical Clinic of the Sylvanus Olympio University Hospital in Lome, International Journal of Internal Medicine, Vol. 9 No. 1, 2020, pp. 20-22. doi: 10.5923/j.ijim.20200901.03.
1. Introduction
- Sharp syndrome or mixed connective tissue disease (MCTD) is the association in a patient of symptoms identical to at least two connective tissue disease [1]. As a reminder, the group of connective tissue disease includes autoimmune diseases that have in common a diffuse, inflammatory, chronic involvement of the collagen fibers of the connective tissue [1]. MCTD was first described in 1972 by Gordon Sharp and his colleagues as an autonomous clinical-biological portrait, associating manifestations belonging at the same time to systemic lupus erythematosus, systemic scleroderma, polymyositis and to rheumatoid arthritis [2]. This syndrome is associated with the presence in the blood of antinuclear antibodies with speckled fluorescence whose antigen corresponds to a ribonucleoprotein [2]. Other autoantibodies have been described [3]. Sharp syndrome is rare in black Africans. This is evidenced by the rare series described [4,5,6]. To date, no case has been described in Togo [7]. In this work, we report a case of Sharp syndrome diagnosed in a 39-year-old patient at the Sylvanus Olympio University Hospital in Lomé.
2. Case Report
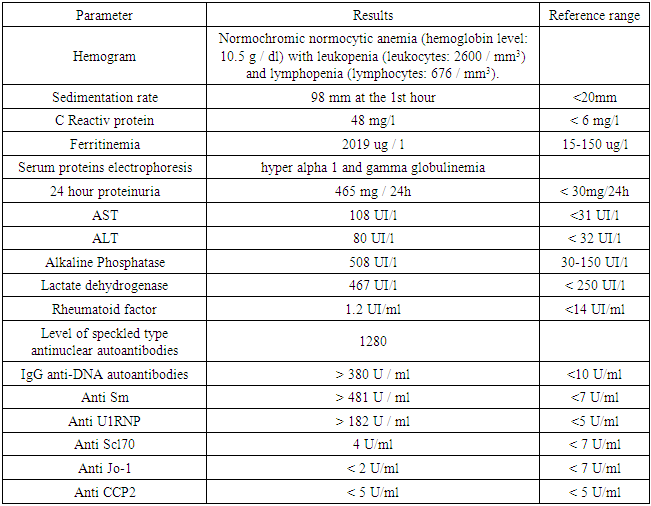
- She is a 39-year-old patient, with no particular pathological history, admitted for polyarthralgia that has been progressing for 5 months. The onset of symptoms dates back to 05 months prior to admission marked by the installation of polyarthralgia with inflammatory schedule affecting bilaterally the knee, elbow and wrist joints in a febrile context. These affected joints were the site of swelling. The associated signs were amenorrhea for 5 months, hair loss, dry cough with a nasal discharge and a deterioration of the general condition. She was admitted to the medical-surgical clinic of the Sylvanus Olympio University Hospital in Lome after several unsuccessful outpatient consultations. Entrance examination noted a deterioration in the general condition (weight loss, asthenia, and anorexia), discrete edemas of the lower limbs, a clinical anemic syndrome, polyarthritis affecting bilaterally the joints of the elbow, knee and wrist with relative functional impotence, brittle hair, photosensitization to exposed areas, myalgias. There was no Raynaud’s phenomenon. Neurological, digestive, gynecological exams were normal.The hemogram noted a normochromic normocytic anemia with leukopenia and lymphopenia. A pregnancy test performed was negative. A biological inflammatory syndrome was noted. Serologies for HIV, viral hepatitis and syphilis were negative. 24 hour proteinuria was elevated. There was a syndrome of cytolysis and hepatic cholestasis. Blood ionogram, glycemia, thyroid checkup, chest X-ray, electrocardiogram, and cardiac Doppler ultrasound were normal. The level of speckled type antinuclear autoantibodies was 1280 (16 × N); IgG anti-DNA autoantibodies were greater than 380 U / ml (38 × N); anti Sm greater than 481 U / ml (70 × N); anti U1RNP greater than 182 IU / ml (36 × N) (Table 1). We retained the diagnosis of mixed connective tissue disease or SHARP syndrome using the diagnostic criteria of Alarcon-Segovia [8] and the JMHW (Japanese Ministry of Health and Welfare) [9]. The patient was treated with analgesics and corticosteroid therapy (injectable prednisone 2 mg / kg / day for 10 days), followed by prednisone tablets. This treatment resulted in a progressive regression of the symptoms. She was discharged from hospital with prednisone tablet and supplemented with calcium and vitamin D. She was readmitted to the hospital 2 months later for dermohypodermitis which progressed well under antibiotic treatment and local care.
|
3. Discussion
- Mixed connective tissue disease is an autoimmune disorder of unknown etiology first described in 1972 by Sharp who noted that patients with high-titre of anti-ribonucleoprotein autoantibodies (RNP) clinically presented characteristic manifestations of both connective tissue diseases such as scleroderma, systemic lupus erythematosus or polymyositis or rheumatoid arthritis [8]. Mixed connective tissue disease can be defined as the presence of antibodies directed against the ribonucleoprotein complex in the plasma, but also of other discovered antibodies [3]. The prevalence of mixed connective tissue disease in patients with a high titre of anti RNP antibodies is about 70% [10].From an epidemiological point of view, this entity is rare in the black Africans in sub-Saharan Africa, as shown by the rare series described: 07 cases in Gabon, 03 cases in Senegal [4], 02 cases in Ivory Coast [5]. A prevalence of the female sex has been found. The mean age of the patients was 39.5 years with extremes at 18 and 56 years and the mean duration of symptoms before the diagnosis of MCTD was 16.5 months with delays ranging from 2 months and 6 years [6]. Our case also involved a 39-year-old black African woman with a progression time of approximatively 05 months.There is a clinical polymorphism in mixed connective tissue diseases. To facilitate the diagnosis, diagnostic criteria have been established. These include the diagnostic criteria of Alarcon-Segovia villareal 1976, reviewed in 1987 [8], the criteria of SHARP 1987 [2], the criteria of JMHW 1987 [9]. Studies have been carried out to determine the sensitivity as well as the specificity of diagnostic criteria such as those of Doria [11] and Amigues [12]. It was found that the Alarcon-Segovia and JMHW criteria were the most widely used and the Sharp criteria were less used. The Alarcon-Segovia classification was simple and therefore was more beneficial for classifying mixed connective tissue disease in general while the JMHW criteria were more suitable for an analysis of each sign and symptom of mixed connective tissue disease [9]. In our case, the diagnosis of mixed connective tissue disease or SHARP syndrome was evoked on the basis of diagnostic criteria of Alarcon-segovia and confirmed by those of JMHW.The clinic is dominated in our series by polyarthritis, brittle hair, photosensitization to uncovered areas, myalgia. There was no Raynaud’s phenomenon. Raynaud's phenomenon seems to be rare in the MCTD of black African subjects [6]. Raynaud's phenomenon is at the forefront of the disease and often the first in a series reported in India [13]. In the Norwegian MCTD registry, it is the most consistent manifestation after the anti-U1RNP positivity [14]. Arthralgia is a constant sign in MCTD hence the rheumatological interest of the disease in the recent American cohort of Ungprasert P, arthralgia was the first manifestation followed by Raynaud's phenomenon and puckered fingers in 86%, 80% and 64% of cases respectively at the time of diagnosis [15]. The frequency of myalgia appears to be higher in African MCTDs [11] compared to Caucasian MCTDs in America and Europe where it varies between 24% and 42% [14].Prognostically, the evolution of mixed connective tissue disease is marked by complications that can lead deaths. The causes of mortality in MCTDs are: pulmonary arterial hypertension (26.1%), respiratory failure (23.2%), heart failure (15.9%), infections (10%), renal failure (5.8%), malignancy (4.4%), digestive hemorrhages (2.9%) [16]. In the Gabonese series, 01 deaths were recorded out of the 07 cases (mortality rate of 14.3%), death related to pulmonary arterial hypertension [6]. The only complication noted in our patient was dermohypodermitis, which progressed well under treatment.From a therapeutic point of view, there is no consensual scheme particularly recommended in MCTD to date [6]. L. Missounga used prednisone in all of their patients in combination with methotrexate in 71.4% of the cases; azathioprine was used in 14.3% and the combination of methotrexate and hydroxychloroquine in 42.8% of the cases [6]. In our case prednisone was the main arsenal of our therapy with a good prognosis.
4. Conclusions
- SHARP syndrome is a rare condition, as evidenced by the small series of cases reported to date. Diagnosis is based on clinical and paraclinical criteria. The evolution can be interspersed with various complications that can affect the short, medium and long term prognosis.