-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Genetic Engineering
p-ISSN: 2167-7239 e-ISSN: 2167-7220
2024; 12(2): 13-16
doi:10.5923/j.ijge.20241202.01
Received: Feb. 1, 2024; Accepted: Feb. 25, 2024; Published: Mar. 11, 2024

Estimation of the Y-Chromosomal Short Tandem Repeat Mutation Rates in Case of DNA Expertise Practice
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLSardarkhodja Karimovich Kurganov1, 2
1Republican Centre for Forensic Expertise, Tashkent, Uzbekistan
2Institute of Biophysics and Biochemistry at the National University of Uzbekistan, Tashkent, Uzbekistan
Correspondence to: Sardarkhodja Karimovich Kurganov, Republican Centre for Forensic Expertise, Tashkent, Uzbekistan.
| Email: |  |
Copyright © 2024 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

In the practice of forensic genetic examination, there are cases when an unknown male person leaves the biological trace on physical evidence. In such cases, along with the definition of the genotype of an unknown person, studies on the Y chromosome are performed on the STR loci of nuclear DNA. In most cases, these studies lead to a successful disclosure of the crime. But sometimes, despite the large number of samples examined, the suspect cannot be identified. In most such cases, DNA samples of individuals living in the same area are examined. To narrow the circle of suspects, calculate the lifetime of a common ancestor, through which it is possible to obtain information about related affinity and narrow the circle of suspects. Later, these results will be used to determine the relationship on the paternal line, where a reliable knowledge on mutation properties is necessary for correct data interpretation. When determining the degree of solution on the paternal line, if discrepancies between the child's father and other paternal relatives are not taken into account, population-specific mutation rates should be used to determine if this is a mutation or a true exception. Therefore, in this study, we aim to determine the mutation rates of 17 Y-STR loci in Uzbekistan.
Keywords: Y-STR, DNA analysis, Mutation, Y chromosome
Cite this paper: Sardarkhodja Karimovich Kurganov, Estimation of the Y-Chromosomal Short Tandem Repeat Mutation Rates in Case of DNA Expertise Practice, International Journal of Genetic Engineering, Vol. 12 No. 2, 2024, pp. 13-16. doi: 10.5923/j.ijge.20241202.01.
Article Outline
1. Introduction
- The human Y chromosome represents about 2% of the total human genome and is approximately 60 Mb in length [1,14]. Most of the Y chromosome consists of a non-recombinant region of the Y chromosome (NRY) [11]. The NRY is inherited intact through paternal lineages unless mutation/s have occurred. Because of such inheritance pattern, STR markers located in the NRY region have become useful for applications including genetic structure studies, paternity testing, identification of disasters male victims, identification of male lineages for anthropology purposes, and the identification of male perpetrators in sexual assault criminal cases [4,5,6,10,13]. The first use of STR on the Y chromosome occurred in 1992 and this STR is now known as DYS19. Since then the potential use of Y-STR analysis for forensic casework has been recognized and well documented. Y-STRs, which have an average mutation rate of about 10−3 per locus per generation [2], have proven to be useful for forensic applications and have been included in several commercial Y-STR kits.Although the greatest value of Y STRs is male specificity, this also turns into a major limitation due to the existence of an identical haplotype within a male lineage [9,15]. This means that while currently used Y-STRs are able to reliably differentiate between different male lineages, they cannot resolve these lineages down to individual level in case of paternal relatives [3]. When determining the degree of solution on the paternal line, if discrepancies between the child's father and other paternal relatives are not taken into account, population-specific mutation rates should be used to determine if this is a mutation or a true exception. Therefore, in this study, we aim to determine the mutation rates of 17 Y-STR loci in Uzbekistan.
2. Materials and Methods
2.1. Objects of the Research
- The subjects of the study were blood samples and dried saliva on sterile gauze tampons, selected from 1.170 individuals.
2.2. Samples Collection
- Samples were collected from people living in the districts of Chirchik (200 unrelated males), Angren (150 unrelated males), Zangiota (100 paternal relatives) and Kibray (120 paternal relatives), also samples were collected from 300 (fathers and sons) pairs unrelated males throughout Uzbekistan.
2.3. DNA Extraction
- Genomic DNA was extracted from peripheral blood and dried saliva samples using the salting-out method [8].
2.4. DNA Quantification
- After isolation, the quantity of genomic DNA of each sample was determined by quantitative real-time polymerase chain reaction (PCR) using the Quantifiler™ Human Male DNA Quantification kit (Thermo Fisher Scientific), which includes internal positive control to test for the presence of PCR inhibitors in the DNA extracts. Quantitative real-time PCR was performed on 7500 Real-Time PCR System (Applied Biosystems).
2.5. PCR amplification and Detection
- To ensure successful amplification, 0.5 ng to 1 ng of DNA was used for each multiplex amplification reaction. All thermal cycling was conducted on Applied Biosystems® GeneAmp® PCR System 9700 thermal cyclers. PCR amplification using Y-filer PCR Amplification Kit (Thermo Fisher Scientific) was performed as recommended by the manufacturer, although half of the recommended reaction volume (12.5 μl) was used. Separation and detection of the 17 Y-STR loci were performed using the 3130xl Genetic Analyser (Applied Biosystems) 16-capillary array system and filter set G5. Each sample was prepared by adding 1 mL PCR product to 14 mL of Hi-DiTM formamide and 0.4 mL GeneScanTM-500 LIZTM internal size standard (Thermo Fisher Scientific). The sample run data were analyzed, together with an allelic ladder and positive and negative controls, using GeneMapper ID-X v3.2 (Applied Biosystems) software.
2.6. Statistical Analysis
- Comparison information of the sample data was generated using an in-house software program involving DNA-expert macros designed to check for allele sharing across all loci. Obtained mutations were compared with those available at the YHRD (Y-Chromosome Haplotype Reference Database) [16].
3. Results and Discussion
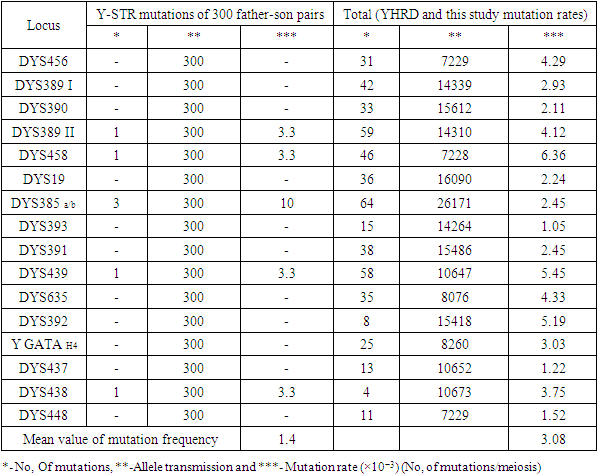
- We assessed the mutation rates of 17 Y-STR loci in Uzbek population. Samples of 300 father-son pairs from all geographic regions of Uzbekistan were typed, and each Y-STR locus pair was compared.We observed a total of seven mutations (DYS389 II, DYS458, DYS385 a/b (three in DYS385 a/b locus), DYS439 and DYS438). The highly polymorphic Y-STR locus DYS385 was observed to have a higher mutation rate compared to all other Y-STRs loci analysed. In this study, the observed higher specific locus mutation rate for Y-STR locus DYS385a/b (if treated as single locus) was 1.0×10-2. Among the three mutations, two repeat losses and one repeat gain were observed. The mutation rates of this study were compared with those available at the YHRD (Table 1). It was found that the DYS438 (3.75×10−3), DYS458 (6.36×10−3) and DYS389 II (4.12×10−3) loci had the highest mutation rate in both YHRD and in our study (Table 1). A low mutation rate was obtained in DYS385 a/b (2.45×10−3) and DYS439 (0.38×10−3) loci.
|
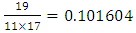 (Mutations per haplotype)In this sample study, the average observed number of mutations per marker is 0.101604.Now we can approximately estimate the age of the common ancestor. The rate of mutations for our 17-marker haplotype: 0.031 mutations per haplotype, or 0.0018 mutations per marker. The duration of one generation is assumed to be 25 years [17].The age of a common ancestor is obtained by dividing the average observed number of mutations per marker by the rate of mutations (also on the haplotype):
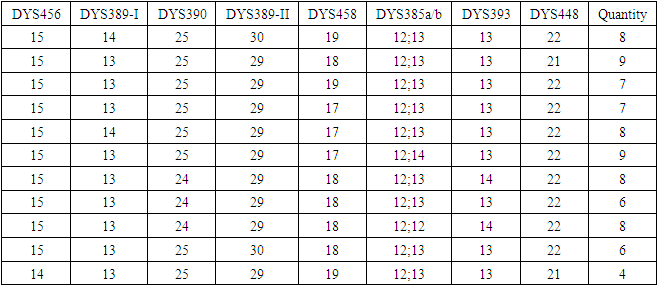
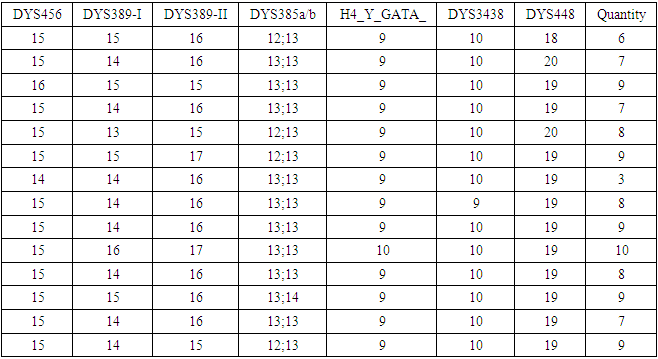
(Mutations per haplotype)In this sample study, the average observed number of mutations per marker is 0.101604.Now we can approximately estimate the age of the common ancestor. The rate of mutations for our 17-marker haplotype: 0.031 mutations per haplotype, or 0.0018 mutations per marker. The duration of one generation is assumed to be 25 years [17].The age of a common ancestor is obtained by dividing the average observed number of mutations per marker by the rate of mutations (also on the haplotype):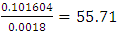 generations.Multiplying 55.71 generations by the duration of one generation of 25 years, we obtain 1392 years. Since the calculation is approximate, we recommend rounding the result down to tens of years. In our case, rounding gives 1390 years. 1390 years is an approximate, rough estimate of the age of a common ancestor.The same method was used to investigate the genetic set of 200 people living in the town of Chirchik. Among this random sample, 109 people were related on the paternal line. Among them, when studying a set of loci with a high frequency of mutations, 24 individual mutations were found (Table 3).The total number of mutations per sample (24 mutations) is divided by the number of haplotypes (14 haplotypes) and the number of markers in the haplotype (17 markers).
generations.Multiplying 55.71 generations by the duration of one generation of 25 years, we obtain 1392 years. Since the calculation is approximate, we recommend rounding the result down to tens of years. In our case, rounding gives 1390 years. 1390 years is an approximate, rough estimate of the age of a common ancestor.The same method was used to investigate the genetic set of 200 people living in the town of Chirchik. Among this random sample, 109 people were related on the paternal line. Among them, when studying a set of loci with a high frequency of mutations, 24 individual mutations were found (Table 3).The total number of mutations per sample (24 mutations) is divided by the number of haplotypes (14 haplotypes) and the number of markers in the haplotype (17 markers).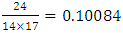 (mutations per haplotype)In this sample study, the average observed number of mutations per marker is 0,10084.
(mutations per haplotype)In this sample study, the average observed number of mutations per marker is 0,10084.
|
|
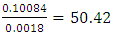 generations.Multiplying 55.30 generations by the duration of one generation of 25 years, we obtain 1382 years. Since the calculation is approximate, we recommend rounding the result down to tens of years. In our case, rounding gives 1380 years. 1380 years is an approximate, rough estimate of the age of a common ancestor.
generations.Multiplying 55.30 generations by the duration of one generation of 25 years, we obtain 1382 years. Since the calculation is approximate, we recommend rounding the result down to tens of years. In our case, rounding gives 1380 years. 1380 years is an approximate, rough estimate of the age of a common ancestor.4. Conclusions
- The current knowledge about the Y chromosome and the availability of markers with divergent mutation rates make it possible to answer questions on relatedness levels which differ in time depth; from the individual and familial level to the surnames, clan and population level. The present study shows the locus specific mutation rate estimate of 17 Y-STRs loci. Considering the results obtained in the present study, it can be concluded that, the reliable knowledge about mutation rates of 17 Y-STRs loci used in forensics and paternity testing in-volving males is very important for a correct interpretation of results.
Ethical Approval
- This study was reviewed and approved by the ethics of the collective council of the Republican Center for Forensic Expertise (Republic of Uzbekistan).