-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Genetic Engineering
p-ISSN: 2167-7239 e-ISSN: 2167-7220
2019; 7(2): 19-24
doi:10.5923/j.ijge.20190702.01

MYH7 Gene Mutations in Dilated Cardiomyopathy Patients: About 190 Cases at the Institute of Cardiology, Abidjan
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLFatoumata Koné1, 2, Carine Yapo -Yao1, Thomas Toni2, Cackouoh Carole Constance Koudou1, Fabienne Armande Kouakou1, Bénédicte Dakouri Koné3, Roland N’Guetta3, Marie-Laure Attoungbré Hauhouot1, 3
1Department of Biochemistry and Molecular Biology, Faculty of Pharmaceutical and Biological Sciences, FHBU –Abidjan, Ivory Coast
2Centre de Diagnotic et de Recherche sur le SIDA et les Autres Maladies Infectieuses, Ivory Coast
3Institut of Cardiology of Abidjan, Ivory Coast
Correspondence to: Fatoumata Koné, Department of Biochemistry and Molecular Biology, Faculty of Pharmaceutical and Biological Sciences, FHBU –Abidjan, Ivory Coast.
| Email: |  |
Copyright © 2019 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Cardiomyopathies are one of the main causes of heart failure and dilated cardiomyopathy (DCM) is the most common among them in Africa. Several genetic mutations are involved in the etiology of DCM. Thus, the objective of this study was to look for mutations of the MYH7 gene in patients suffering from DCM through a cross-sectional study. For all patients recruited, we collected the socio-demographic, clinical, therapeutic and radiological data. A tube of venous blood was taken in order to research by sequencing mutations on exons 13, 24 and 31 of the MYH7 gene. The study population included 190 patients. They were predominantly male (72.00%) and their mean age was 55.18 ± 13.83 years. The major personal medical history was high blood pressure (20.00%). A family history of heart disease and sudden death was found in 16.80% and 12.60% of patients, respectively. All patients benefiting from sequencing (n = 32) had at least one mutation in the MYH7 gene, so a prevalence of 100%. A total of 29 mutations were observed, divided into 62.07% missense mutations (n = 18/29), 24.14% silent mutations (n = 7/29), 10.34% deletions (n = 3/29) and 3.45% insertions (n = 1/29). Exon 24 was the most unstable with 21 mutations and some patients had multiple mutations. Mutations in the MYH7 gene were frequent and diverse in patients with DCM in Côte d'Ivoire. It is necessary to establish the link between these mutations and their importance in early management of DCM.
Keywords: MYH7, Mutations, Dilated cardiomyopathy, Heart failure
Cite this paper: Fatoumata Koné, Carine Yapo -Yao, Thomas Toni, Cackouoh Carole Constance Koudou, Fabienne Armande Kouakou, Bénédicte Dakouri Koné, Roland N’Guetta, Marie-Laure Attoungbré Hauhouot, MYH7 Gene Mutations in Dilated Cardiomyopathy Patients: About 190 Cases at the Institute of Cardiology, Abidjan, International Journal of Genetic Engineering, Vol. 7 No. 2, 2019, pp. 19-24. doi: 10.5923/j.ijge.20190702.01.
Article Outline
1. Introduction
- Cardiomyopathies lead for heart failure (HF) and the most common form in African is dilated cardiomyopathy (DCM) [1]. In developed countries, it is a major cause of heart transplantation. In Africa, the prevalence of DCM is around 5 to 20% [2, 3] and it is responsible of frequent hospitalization whith 17-48% for heart failure [2, 4]. The advent of echocardiography has really improved the diagnosis, but it remains limited for the etiological diagnosis [5] So, the etiology remains mostly unknown, leading to difficulties of adaptation and early treatment management [6, 7]. Idiopathic DCM account for more than 30% of diagnosed DCM [3, 8, 9]. More than 30 genes have been identified in the etiology of primary DCM and the most common are the LMNA gene coding for lamin A/C, the MYH7 gene coding the myosin heavy chain, the TNNT2 gene coding for troponin T. Lamin, beta-myosin and troponin are proteins of the cardiomyocyte which is the structural units of the myocardium. Changes in their synthesis may cause a failure of myocardial function, thus contributing to the pathogenesis of DCM [10, 11]. The aim of this study was to look for the mutations of the MYH7 gene in patients with DCM in Ivory Coast.
2. Material and Methods
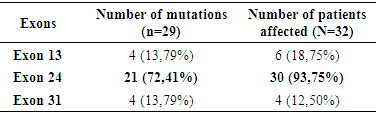
- This work was carried out through a prospective descriptive study from August 2014 to January 2018 at the Institute of Cardiology of Abidjan (ICA). The study population was selected based on 2 conditions. First group: all patients who have been admitted to the ICA emergency unit and diagnosed with dilated cardiomyopathy according to ultrasound, i.e., left ventricular dilatation (Left Ventricular diameter> 55mm) and a decrease in the left ventricular ejection fraction (Ejection Fraction <45%). Those ones have to do the coronarography test; they were retained in the study of the MYH7 genes if the coronarography was normal. Second group: patients known as having DCM and followed at the ICA with a normal coronary angiography result. To be included, all participants meeting this first condition, gave their consent to participate in the study. The criteria for non-inclusion were to not have dilated cardiomyopathy or to have refused to sign the consent form. The study was conducted with the authorization from the board of staff of the Institute of cardiology of Abidjan. Therefore, the patient’s identity was not available for others because they were designed by an anonymous system particular for this study.After obtaining informed consent from patients, we used survey cards to record socio-demographic, clinical and para-clinical parameters. In addition, we collected, venous blood for the search of mutations on exons 13, 24 and 31 of the MYH7 by sequencing. The choice of this gene is due to the fact that it is one of the genes whose mutations are frequently found in patients with DCM [12, 13, 14]. In the search for genetic mutations, we took 3mL of venous blood and after centrifugation; we used the pellet of blood cells for the analysis. We carried out the DNA extraction on this pellet according to the Invitrogen kit method. Then the concentration and purity of the DNA of the extracts were measured by spectrophotometery (BioPhotometer Plus®). The extraction was resumed when the DNA concentration was <30 ng / μL and / or the purity <1.5. After the validation of the quality of the extracted DNA, we carried out the amplification of the interested DNA fragments by conventional polymerase chain reaction (PCR). The primers used were as following [14]: exon 13 Forward: TTACAGGCATGAACCACACACC-Reverse: GTGAACTTGAAAACTCTCATCCC; exon 24: Forward: GACCATACTGACCTTGACCC-Reverse: AGACATGGCATATCTAGGCC; exon 31: Forward: ATCCTCCCCACCCTCTGC-Reverse: GAGGATGGCTCTTGGCCTCT.For each PCR test, the reaction mixture consisted of 50 μL at the rate of 5 μL of 10X buffer, 3 μL of magnesium chloride (2.5 mM), 4 μL of each of the dNTP (100 mM), 4 μL of each of the forward and reverse primers (10 mM), 0.3 μL of Taq polymerase (1 IU / L), 12.7 μL of water grade molecular biology and 5 μl of DNA extract (≥30 ng / μL). We performed 3 single-target PCR (not multiplex PCR) and all of the PCR programs consisted of an initial denaturation at 94°C for 3 minutes (min), 40 cycles of (denaturation at 95°C for 30 seconds (s), hybridization at 60°C for 30 s and elongation at 72°C for 2 min) followed by a final elongation at 72°C for 7 min followed by a hold at 4°C [14]. After PCR, the search for mutations was performed by sequencing of the 3 DNA fragments of interest. There were some non-specific amplifications and bad sequences, so some sequences was not analysed. The flowchart for the number of sequences analyzed is presented in Figure 1. However, we considered the sequences analysed as representative of the 32 samples sequenced. We used 2 MYH7 gene reference sequences: the reference DNA sequence from Genbank with the accession number: NG_007884 [15] for the confirmation of the exons’s localization and the mRNA’s cDNA complete coding sequence also from Genbank with accession number: BC112173 [16] for the mutations’s reseach. About the reference sequence, [15], MYH7 consists of 29924 bp linear DNA and it has 40 exons. The mutations reported were classified as (i) silent mutation for nucleotide change in MYH7 gene without amino acid change in beta-myosin protein sequence, (ii) missense mutation for nucleotide change in MYH7 gene with amino acid change in beta-myosin protein sequence, (iii) deletion for the loss of nucleotide in MYH7 gene and (iv) insertion for the adding of nucleotide in MYH7 gene.We used Ape and SeqScape_3 bio-informatics software for sequence analysis and SPSS software version 16.0 for statistical data analysis.
3. Results
- This study involved 190 patients, 137 men and 53 women giving a sex ratio of 2.60 and the mean age was 55.18 ± 13.80 years. The major personal antecedent was high blood pressure (20.00%). The family history of cardiac disease and sudden death was found in 16.80% and 12.60% of patients respectively, but no family history of DCM was reported. Cardiomegaly was present in 97.20% of patients.Of the 32 samples sequenced for each exons there were some bad sequences. So, 13 were analyzed for exon 13, 30 for exon 24 and 4 for exon 31 (Figure 1).
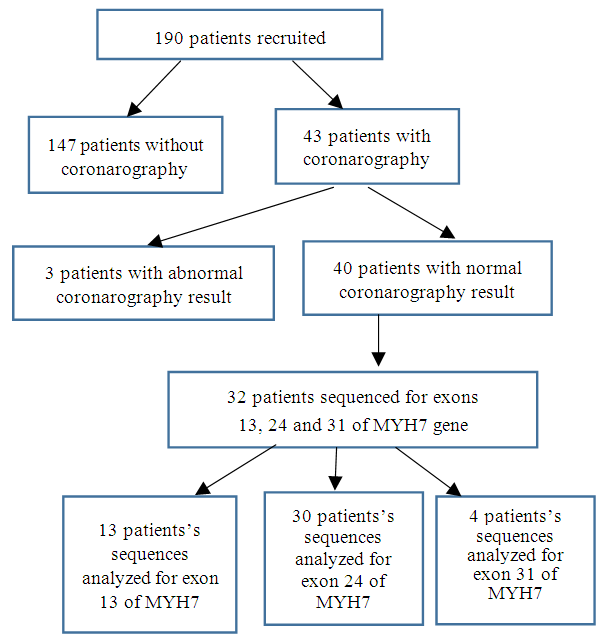 | Figure 1. Flowchart of the effect disaggregation in the study |
|
|
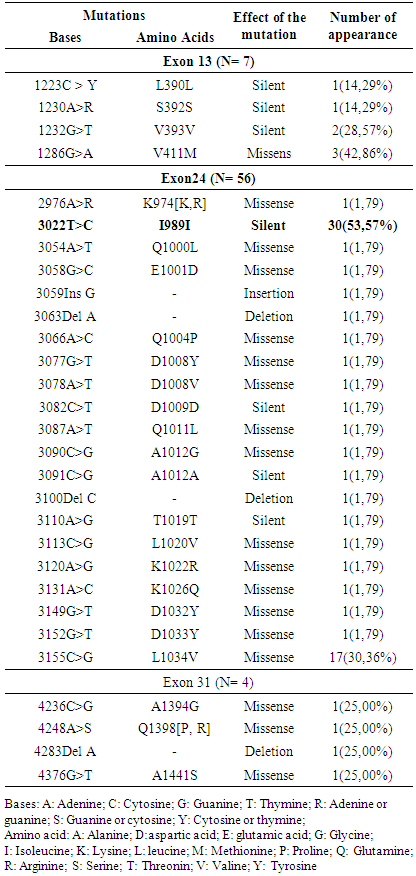
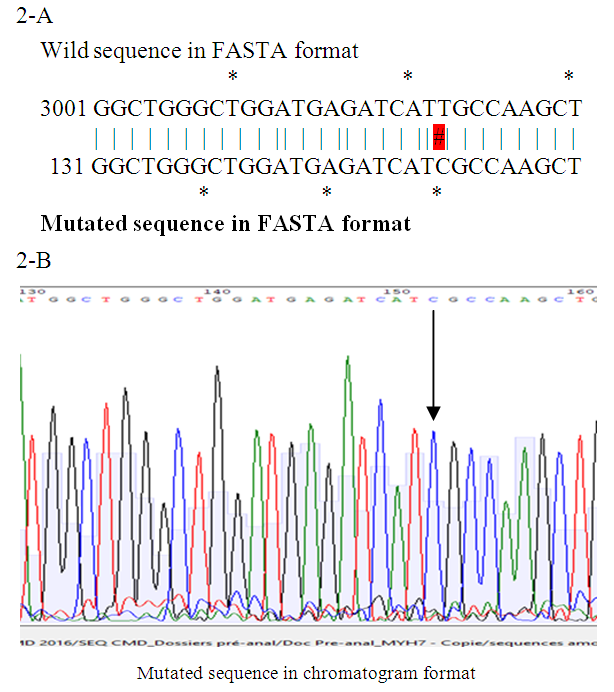 | Figure 2. Sequencing results in FASTA (2-A) and chromatogram (2-B) formats of the 3022T>C mutation in the same patient |
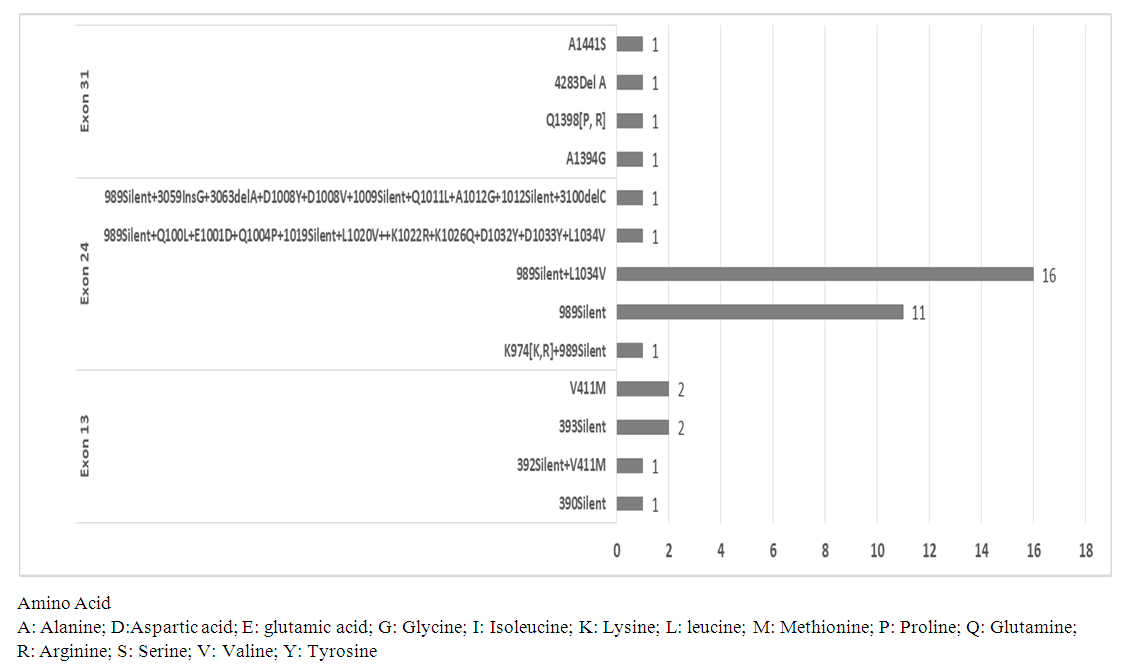 | Figure 3. Distribution of patients according to the type of mutation observed in the 3 exons |
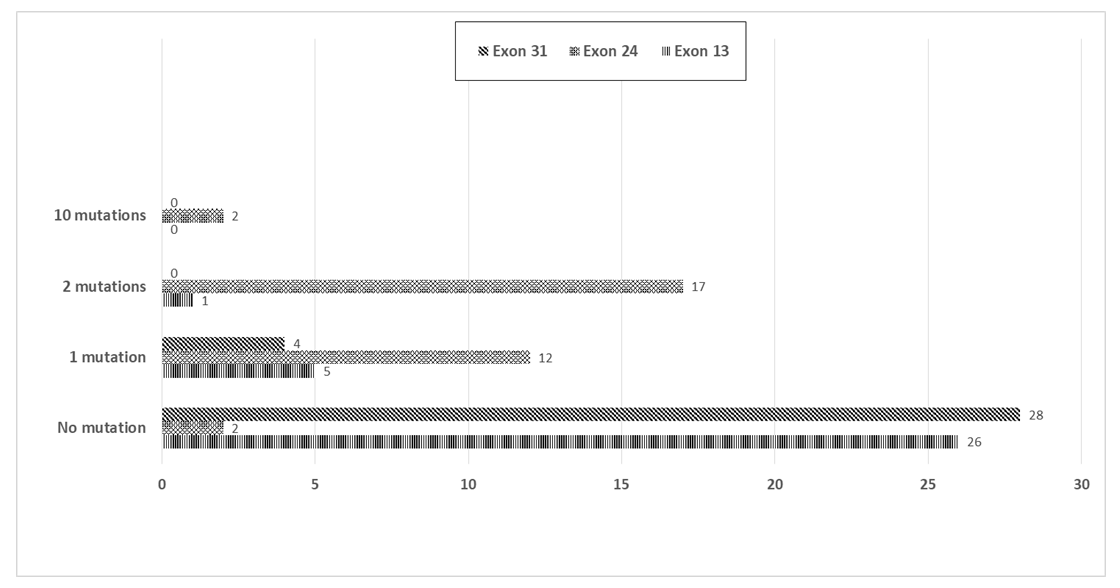 | Figure 4. Distribution of patients according to the number of mutation observed in the 3 exons |
4. Discussion
- From August 2014 to January 2018, we recruited 190 dilated cardiomyopathy patients (137 men and 53 women) whose mean age was 55.18 ± 13.80 years. In these patients we looked for mutations in MYH7 gene. The study showed the presence of various and frequent mutations in dilated cardiomyopathies patients. The distribution of the study population, according to the age and gender reflected that of black adult patients monitored for treatment in cardiology. Regardless of the studies conducted and the disease of interest, the average age of the population generally ranges from 50 to 60 years, with male predominance [9, 10, 17]. The predominance of men affected by this illness is well established: it often affects men 2 to 3 times than women [3].Personal history of cardiovascular diseases was found in which high blood pressure predominated. This presence of cardiovascular diseases story prompted towards the diagnosis of secondary DCM that may or may not be associated with genetic mutations. The family story of heart disease and sudden death reported in this study suggested the presence of hereditary DCM in this study population. Our results were lower than those of Coulibaly [18] who reported 76.70% of high blood pressure history. This difference could be explained by the fact that ICA is a specialized center for the management of cardiovascular diseases, so there could have been a better management of the hypertension. These good care conditions allowed to avoid the occurrence of complications such as DCM. No history of DCM was reported, probably because of the ignorance of this illness by the populations.On MYH7 gene, the 100% prevalence of mutations observed differed from the results found in the literature where the prevalence is estimated to be around 4.00%. Indeed, in the context of developed countries, Millat et al. [13], Hershberger et al. [12] and Villard et al. [14] found prevalences of 3.80%, 4.20% and 7.00% respectively. This difference could be explained by several reasons. On the first hand due to the fact that our population was composed exclusively of black African patients while the previous studies were conducted on caucasian populations. On a 2nd hand, data on the genes involved in DCM in black Africans being almost not available, the reference sequence used for the bio-informatic analysis of the sequences in our study (black african) comes from a population different from that studied (caucasian). On a 3rd hand, this is one of the first genetic study conducted in black african population and other authors showed that the mutions involved in DCM seem to increase with the efforts of descovry [19]. The total number of mutations found was 29 and concerned the 3 exons targeted. Our results were superior to those reported by Millat et al. [13] and Villard et al. [14] who found 4 and 7 mutations respectively. This high number of mutations with a high proportion of silent mutations in our results may contain genetic polymorphism mutations. Polymorphism mutations are nucleotide changes that do not lead for any disease, so we aimed to investigate their disease causing action in further studies. Villard et al. [14] found 30 mutations in the MYH7 gene and these included 23 polymorphism mutations. Also, in the study conducted by Millat et al. [13], the 4 mutations reported were all DCM causing mutations. On the target exons in our study, the mutations previously reported in the literature were unique: the T412N mutation on exon 13, the T1019N on exon 24 and the E1426K on exon 31. We observed none of those mutations in our study population, despite of the high prevalence of mutations and their diversity. These differences could be due to the genetic variability between our study population and those of the literature. In addition, Côte d'Ivoire is a tropical and developing country. So, our study population is exposed to particular environmental conditions leading for the occurrence of genetic mutations [20, 21]. These poor conditions include recurrent microbial infections whose prevalence is usualy high in developing countries. It is established that microbial infections are responsible for significant genetic variability [21].Regarding the type of mutations found, we identified in decreasing order missense mutations, silent mutations, deletions and insertions. These results were similar to those reported by Hershberger et al. [12, 19]. Apart from the silent mutations, others could be involved in the pathogenesis of DCM because they generally lead for the synthesis of modified proteins with different properties compared to the wild protein (missense mutation) or the formation of non-functional truncated proteins (insertions and deletions). In the case of MYH7 gene, such mutated myosin could induce a decrease in the contractile function of the sarcomere and lead as etiology of CMD.We also found combined mutations in the same exon and combined mutations in differents exons. These associations are important for the monitoring of the patients because when some mutations stand alone they play as polymorphism mutation and when they are combined with other mutations, they lead for diseases.
5. Conclusions
- Mutations in the MYH7 gene were frequent and various in patients with idiopathic DCM. Nevertheless, it is necessary to investigate the inheritance of these genetic mutations in families and establish the impact of knowing these mutations in a family on the early management of DCM.
ACKNOWLEDGEMENTS
- To DJAMAN Allico Joseph: 01 BP 490 Abidjan 01; Laboratory of Pharmacodynamy-Biochimiy, UFR of Biosciences (FHB University) & Institut Pasteur of Ivory Coast. He helped us for the DNA sequencing.