-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Genetic Engineering
2012; 2(3): 19-27
doi: 10.5923/j.ijge.20120203.01
Association of Periodontal Diseases with Genetic Polymorphisms
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLMegha Gandhi , Shaila Kothiwale
KLE V K Institute of Dental Sciences, KLE University, Belgaum, Karnataka, India
Correspondence to: Megha Gandhi , KLE V K Institute of Dental Sciences, KLE University, Belgaum, Karnataka, India.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Periodontal diseases are multifactorial in nature. While microbial and other environmental factors are believed to initiate and modulate periodontal disease progression, there now exist strong supporting data that genetic polymorphisms play a role in the predisposition to and progression of periodontal diseases. Variations in any number or combination of genes that control the development of the periodontal tissues or the competency of the cellular and humoral immune systems could affect an individual's risk for disease. A corollary of this realization is that if the genetic basis of periodontal disease susceptibility can be understood, such information may have diagnostic and therapeutic value. This review aims to update the clinician about various genetic polymorphisms associated with periodontal diseases to aid in a better approach to the condition in the future.
Keywords: Periodontal Disease, Gene Polymorphisms, Chronic Periodontitis, Aggressive Periodontitis, Hereditary Gingival Fibromatosis
Article Outline
1. Introduction
- Periodontal disease is an inflammatory illness that represents the main cause of tooth loss in developed countries, with increasing prevalence in the developing world[1]. Periodontitis is defined as “an inflammatory disease of the supporting tissues of the teeth caused by specific microorganisms or groups of specific microorganisms, resulting in progressive destruction of the periodontal ligament and alveolar bone with pocket formation, recession, or both[2]. Changes that occur in the alveolar bone are crucial because the destruction of bone is responsible for tooth loss. Periodontal diseases are multifactorial in nature. While microbial and other environmental factors are believed to initiate and modulate periodontal disease progression, there now exists strong supporting data that genetic and environmental risk factors play a role in the predisposition to and progression of periodontal diseases (Fig 1)[3,4,5].The application of genetic information and technology for the prediction, diagnosis and treatment of periodontal conditions is conceptually compelling. Several features like cytokines, cell-surface receptors, chemokines, enzymes and others that are related to antigen recognition, the immune system, host response, among others, are determined by genetic components; polymorphisms of which may increase the susceptibility of an individual to periodontal diseases. Identifying genes and their polymorphisms can result in novel diagnostics for risk assessment, early detection of disease and individualized treatment approaches[6]. Thus, genetic epidemiology, including knowledge of genetic polymorphisms, holds promise as one of the tools that may contribute to the understanding of periodontal disease (Fig 2).
2. Evidence for the Role of Genetics in Periodontal Disease
- Periodontal research has greatly expanded to elucidate the role of genetics in periodontal disease states. Consequently, there has been great interest in identifying allelic variants of genes that can be used to assess disease risk for periodontal diseases. Reports of genetic polymorphisms associated with periodontal disease are continuously increasing (Fig 3).In the past it was thought that periodontitis would eventually develop in subjects with a longstanding history of poor oral hygiene and gingivitis[7]. Despite this belief, it was recognized that only a portion of the variability of disease in the population could be explained by environmental factors alone. This variation must have been attributable to either unrecognized components of the environment or differences among individuals in their susceptibilities to disease. Because host susceptibility may be defined in terms of genetic variation, the focus of determining disease susceptibility shifted to quantify the genetic aspects of disease. Epidemiologic studies as well as longitudinal clinical studies of the oral microbial flora supported the development of the theory that although microbial and environmental factors are required for periodontitis, the disease may have a genetic background (Fig 4).
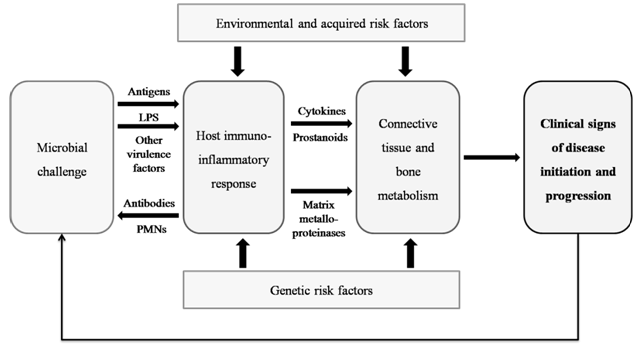 | Figure 1. Model for pathogenesis of human periodontal diseases. (Modified from Page RC, Kornman KS. The pathogenesis of human periodontitis: an introduction. Periodontol 2000 1997; 14: 9-11) |
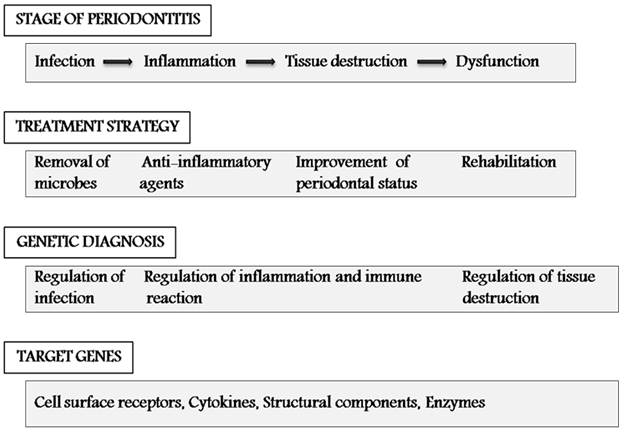 | Figure 2. Relationship between genetic diagnosis and target genes at various stages of periodontitis and their treatment strategy. Each stage of the periodontal disease progression can be assessed based on a genetic diagnosis and treatment can be aimed for the target genes expressed at that stage of disease. (Modified from Takashiba S, Naruishi K. Gene polymorphisms in periodontal health and disease. Periodontol 2000 2006; 40: 94–106) |
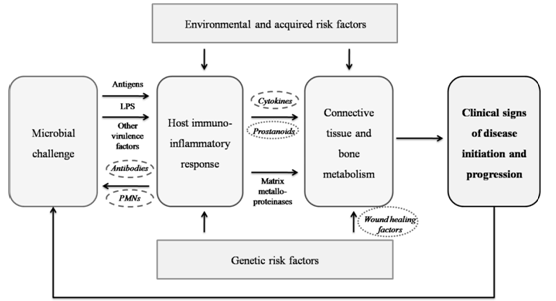 | Figure 3. Genetic factors in periodontitis and their potential biologic influence. Candidate genetic factors with definite influence on disease states (shown in dashed circles); potential factors with inadequate data to prove association with disease (shown in dotted circles) |
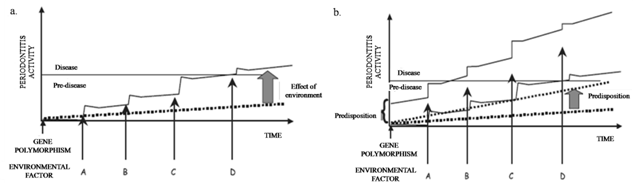 | Figure 4. Effect of multiple assaults of environmental factors on disease progression and the effect of increased predisposition caused by genetic factors a. Periodontitis susceptibility may be regulated by a gene polymorphism (dotted line) without ever resulting in developing overt disease. Hence, periodontitis symptoms might be unaffected by a gene polymorphism. However, with multiple effects of environmental factor assaults, the disease may become apparent (solid line from factors A-D). b. Increased predisposition towards periodontal disease may also result as a function of the basal predisposition (dotted line). Hence, higher “predisposition” leads to increased disease activity. as a result of these effects, rate of disease progression (elbow-shaped line) rises significantly following each environmental assault. (Modified from Takashiba S, Naruishi K. Gene polymorphisms in periodontal health and disease. Periodontol 2000 2006; 40: 94–106) |
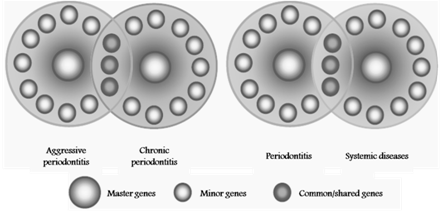 | Figure 5. Possible concept for common polymorphisms. Both chronic and aggressive periodontitis may have shared susceptibility genes, in the same manner as periodontitis may share susceptibility genes with other complex, inflammatory or systemic diseases. (Modified from Yoshie H, Kobayashi T, Tai H, Galicia JC. The role of genetic polymorphisms in periodontitis. Periodontol 2000 2007; 43:102–132) |
3. Periodontal Diseases Influenced by Genetic Polymorphisms
- Given the complex etiology and pathogenesis of periodontal diseases, variations in any number or combination of genes that control the development of the periodontal tissues or the competency of the cellular and humoral immune systems could affect an individual's risk for disease. Evidence for a genetic influence on periodontal diseases comes from multiple sources including familial aggregation and formal genetic studies of aggressive periodontitis, the association of periodontitis with certain Mendelian inherited diseases, and twin studies of chronic periodontitis (Fig 5)[14].
3.1. Chronic Periodontitis
- Chronic periodontitis has been defined as an infectious disease resulting in inflammation within the supporting tissues of the teeth, progressive attachment loss, and bone loss. It is associated with the accumulation of plaque and calculus and generally has a slow to moderate rate of disease progression, but periods of more rapid destruction may be observed. The disease also may be described by the severity of disease as slight, moderate or severe based on the amount of clinical attachment loss[2]. Although no clear genetic determinants have been described for patients with chronic periodontitis, a genetic predisposition to more aggressive periodontal breakdown in response to plaque and calculus accumulation may exist[15]. Also, clinicians have long suspected that susceptibility to periodontitis differs among racial and ethnic groups.Investigations have utilized family studies of probands with chronic (adult) periodontitis or younger subjects with mild/incipient periodontitis. The results suggested that there may be a genetic basis for the less severe forms of periodontitis[16]. The twin model is probably the most powerful method to study genetic aspects of periodontal diseases. The largest twin study included 4908 twin pairs of which, on the basis of questionnaire data, 349 (116 MZ and 233 DZ) pairs reported a history of periodontal disease in one or both pair members. The concordance rates ranged from 0.23 to 0.38 for MZ twins and 0.08 to 0.16 for DZ twins[17]. Michalowicz and co-workers evaluated the periodontal condition (attachment loss, pocket depth, gingival index and plaque index) of 110 adult twin pairs with a mean age of 40 years. The results indicated that between 38% and 82% of the population variance for these measures may be attributed to genetic factors[18]. In a subsequent study the authors estimated that chronic (adult) periodontitis hadapproximately 50% heritability, which was unaltered following adjustments for behavioural variables including smoking and utilization of dental care[19]. In epidemiological studies in the Dutch population, it has been suggested that periodontitis aggregates in families[20,21].Kornman et al reported that a “composite” IL-1 genotype consisting of at least one copy of the more rare allele at both an IL-lα and IL-1β loci was associated with severe periodontitis[22]. Karimbux et al (2012), in their meta-analysis, reported that IL1A and IL1B genetic variations are significant contributors to chronic periodontitis in Caucasians[23].From both the twin studies and familial studies it can be concluded that the basis for familial aggregation of periodontitis appears to be genetic rather than bacterial, environmental or behavioral in nature. The heritable component for periodontitis was not associated with behaviors such as smoking, utilization of dental care and oral hygiene habits. This implies that genes controlling biologic mechanisms, and not behaviors, mediate the genetic influence on disease.
3.2. Aggressive Periodontitis
- Aggressive periodontitis was previously classified as early onset periodontitis (EOP). EOP included prepubertal, juvenile and rapidly progressive forms of periodontitis. Aggressive periodontitis is characterized by rapid rate of disease progression seen in an otherwise healthy individual, an absence of large accumulations of plaque and calculus and a family history of aggressive disease suggestive of a genetic trait. Although diagnoses are based on clinical and radiographic criteria, the immunologic and microbiologic profiles can vary substantially within the subforms[1].Marked aggregation of aggressive periodontitis within families is consistent with a genetic predisposition to this disease. In a United States survey of 7447 dentate individuals aged 13 years and older, the prevalence of severe disease was low in younger age groups. The prevalence of loss of attachment of ≥ 5 mm was 0.3% among 13–17 and 18–24 year olds, and 6.4% among 25–34 year olds in a racially mixed population. In contrast, 40–50% of siblings in families with aggressive periodontitis were similarly affected[24]. One of the largest and most comprehensive segregation analysis for aggressive periodontitis has been performed by Marazita et al (1994). It was suggested by the authors that the most likely mode of inheritance was autosomal dominant in both African-American and Caucasian kindred, with 70% penetrance in African-Americans and 73% in Caucasians [25]. However, familial patterns of disease may reflect not only a common genetic background but also exposure to common environmental factors. Studies have also been conducted on linkage analysis of candidate regions which lie within or near genes that code for enzymes or regulatory molecules that are likely to be involved in the pathogenesis of disease. The human leukocyte antigens (HLA) have been considered to be candidate markers for aggressive periodontitis because they are involved in regulating immune responses. Two antigens that appear to be consistently associated are HLA-A9 and –B15[26]. The risk of disease in subjects with HLA-A9 or –B15 is about 1.5 to 3.5 times greater than in those lacking these antigens. In contrast, the HLA-A2 appears to be less prevalent in aggressive periodontitis patients than controls, suggesting that this antigen somehow may be protective[27]. An IL1B allele has been reported to be in linkage disequilibrium with generalized aggressive periodontitis, suggesting that at least one disease allele is at or very close to this IL1B polymorphism (2q13)[28].Another study concluded that in West European Caucasians, the Asp299Gly TLR4 gene polymorphism was associated with a decreased risk of AgP but not CP. Promoter polymorphisms of the CD14 gene, however, did not influence susceptibility to inflammatory periodontitis in the population cohorts studied[29]. Li et al provided evidence stating FokI polymorphism of vitamin D receptor gene might be associated with generalized aggressive periodontitis in Chinese patients. In addition, the carriage of F allele increases the risk of developing generalized aggressive periodontitis[30].
|
3.3. Monogenic Syndromes
- Aggressive periodontitis has often been reported inpatients with certain monogenic syndromes. These conditions are rare (Table 2). In many of these conditions the permanent dentition is also affected to a greater or lesser extent.The most extensively studied of the monogenic conditions is the Papillon-Lefevre syndrome. It is inherited as anautosomal recessive trait. The classical clinical manifestations of Papillon-Lefevre syndrome are hyperkeratotic lesions of the skin, affecting the palmoplantar surfaces, and aggressive periodontitis, affecting the deciduous and permanent dentitions[31]. The underlying causation of Papillon-Lefevre syndrome has been the subject of considerable debate in the literature. In addition to defects in epithelium formation, reduced lymphocyte response in vitro and decreased chemotaxis and phagocytosis of neutrophils and monocytes have been reported in Papillon-Lefevre syndrome patients. Studies involving genome wide linkage scans of consanguineous families mapped the Papillon-Lefevre syndrome locus to the long arm of chromosome 11, with the gene encoding lysosomal protease cathepsin C[32]. This enzyme is expressed at high levels in many immune cells including polymorphonuclear leukocytes and macrophages and their precursors. In addition, it has been found that cathepsin C is expressed in areas of epithelium often affected by hyperkeratotic lesions such as palms, soles, knees and oral keratinized gingiva[33]. Individuals displaying the Papillon-Lefevre syndrome phenotype are homozygous for the trait. Other relatives who are heterozygotes do not show evidence of either palmoplantar hyperkeratosis or severe periodontitis. The finding that mutations of the cathepsin C gene give rise to Papillon-Lefevre syndrome corroborates earlier suggestions of the cause of this syndrome[31].Another syndrome described by Haim and Munk (1965) has only been observed in the descendents of a religious sect originally from India. This condition displays the same features of hyperkeratosis and severe aggressive periodontitis as Papillon-Lefevre syndrome, with the addition of arachnodactyly and a peculiar deformity of the terminal phalanges[34]. The Haim-Munk syndrome was also found to be linked to the cathepsin C gene. In other words Papillon-Lefevre syndrome and Haim-Munk syndrome are allelic variants of mutations of the cathepsin C gene.
3.4. Refractory Periodontitis
- Patients who respond poorly to periodontal treatment by continuing to lose clinical attachment are said to have refractory periodontitis. Both, aggressive and chronic periodontitis can be refractory to treatment. Neutrophils from refractory disease patients usually display some functional defect. Kobayashi et al tested the association between neutrophil immunoglobulin G receptor (FcγR) polymorphisms and adult (now known as chronic) periodontitis in a Japanese population[35]. These polymorphisms, which reside in genes that encode receptors for the IgG isotypes, correlate with how efficiently PMNs are able to phagocytose opsonized antigens. The frequency of FcγR genotypes did not differ between chronic periodontitis patients and healthy controls. Among patients, however, one allele (FcγRIIIB-NA2) was found to be more prevalent in those who experienced recurrent disease. There have been very few studies to confirm the association between FcγR genotypes and refractory disease.
3.5. Gingival Enlargement
- Gingival enlargement is the overgrowth of the gingiva characterized by an expansion and accumulation of the connective tissue with occasional presence of increased number of cells. It is a form of periodontal disease which can be inherited (hereditary gingival fibromatosis (HGF), associated with other diseases characterizing a syndrome, caused by several factors, such as inflammation, leukemia, etc or induced as an adverse effect of systemic drugs, such as phenytoin, cyclosporine or nifedipine[1]. The gingival enlargement results in both esthetic and functional problems for affected individuals.The inheritance condition in which the gingival tissue spontaneously and progressively enlarges is identified as hereditary gingival fibromatosis (HGF). HGF, previously known as elephantiasis gingivae, hereditary gingival hyperplasia and hypertrophic gingiva, is traditionally considered an autosomal dominant disease. HGF is a rare disease of infancy characterized by progressive gingival enlargement of normal color and firm consistency that is non-hemorrhagic and asymptomatic. Males and females are equally affected at a phenotype frequency of 1:175,000. HGF results in diastemas, malpositioning of teeth, prominent lips and open lip posture. Although the gingival enlargement does not directly affect the alveolar bone, the gingival swelling may increase the bacterial plaque accumulation, inducing periodontitis and bone resorption and halitosis[36].The first case was probably reported by Gross in 1856 and although over the past few years increasing efforts have been made to understand the genetic, molecular and cellular basis of the gingival enlargement of HGF patients, it has been impaired by the intense clinical, genetic and biologic heterogeneity of the disease. Three different loci have been associated with the isolated form of HGF: two mapping to chromosome 2 (GINGF on 2p21-22 and GINGF3 on 2p22.3-p23.3), which do not overlap, and one mappping to chromosome 5 (GINGF2 on 5q13-q22). Of these loci, only the SOS1 (son of sevenless one) gene underlying the GINGF locus has been identified[32]. The SOS1 mutation linked to HGF was observed in a large multigenerational Brazilian family segregating HGF as a highly penetrant autosomal dominant gene. The single insertion mutation, which causes a frame shift and a stop codon resulting in a truncated protein, was segregated in all affected patients and was not identified in unaffected family members or in control subjects[37].Drug-induced gingival overgrowth has been seen in patients following intake of phenytoin, cyclosporine or nifedipine. Phenytoin, cyclosporine and nifedipine are all metabolized by the hepatic cytochrome P450 enzymes. Cytochrome P450 genes exhibit considerable polymorphism, which results in interindividual variation in drug levels. This inherited variation in metabolism of either drug may influence patient serum and tissue concentrations and hence their gingival response[38]. Another genetic marker that has been investigated in relation to drug-induced gingival overgrowth is the human lymphocyte antigen expression (HLA). Investigation of this marker has been confined to the organ transplant patients since their HLA phenotype is determined prior to transplantation. Several studies have reported on the relationship between HLA expression and the incidence of drug induced gingival overgrowth[38,39]. A study reported that patients who expressed HLA-DR1 are afforded some degree of protection against gingival overgrowth whilst HLA-DR2 may increase the development of this unwanted effect[39]. A trend towards an increased presence of HLA-A19 antigen has also been reported although the relationship was not significant after correction for multiple significance testing. To date only HLA-B37 has been identified as a significant risk factor after correction for the effect of multiple significance testing and these patients are protected in some way from the effects of gingival overgrowth. The concept of molecular mimicry in the wider field of periodontal disease or an effect on lymphocyte function have been postulated as the mechanisms that may tie HLA antigens to gingival overgrowth[38].HGF is frequently an isolated disorder, but in some cases, it is associated with other alterations characterizing a syndrome (Table 3)[36].
| ||||||||||||||||||||||||||||||||||||
4. Concluding Remarks
- With recent evidence implicating the role of periodontal disease on systemic health, an understanding of the potential role of genetic polymorphisms on periodontal disease may help in modifying our treatment plan to improve periodontal health, as well as, general health. Emerging data strongly suggest that host genetic factors are important determinants for periodontitis susceptibility. Genetic tests may prove useful for identifying patients who are most likely to develop periodontal disease, suffer from recurrent disease or suffer tooth loss as a result of periodontal disease. Genetic polymorphisms also result in the development of monogenic syndromes of which periodontal diseases are an important sign.The present literature review is the first of its kind to focus on the various diseases affecting the periodontium and how genetic polymorphisms can affect these diseases, unlike previous literature researches wherein the various polymorphisms have been enlisted and discussed. The idea is to instigate the researcher to approach the disease and investigate the possible polymorphisms, when present simultaneously, that could be aggravating or providing protection against the disease, rather than to identify polymorphisms and calculating the possibility of disease causation or prevention individually. At present, the search for risk alleles for periodontitis must focus on candidate gene regions. Genome-wide searches for risk alleles are not feasible due to the limited number of genetic markers currently available for typing. Single nucleotide polymorphisms are likely to be valuable tools in the search for disease alleles. Recently, genome-wide association studies using single nucleotide polymorphisms or microsatellite polymorphisms have become realistic due to the development of promising high-throughput and cost-effective single nucleotide polymorphism typing. As suggested by Tamiya et al, microsatellite-based genome-wide association analysis complemented by end-stage single nucleotide polymorphism typing will be an interesting strategy for genetic dissection of multifactorial pathologies such as common diseases, including periodontitis[40].Future strategies for the utilization of genetic polymorphisms in Periodontics will need to consider at least two factors. The first is to perform large-scale genetic analyses in diverse populations using as many target genes and subjects as reasonably possible. Secondly, there needs to be a development of new statistical analytical methods to combine both genetic and environmental factors.To achieve these goals, multicenter research studies will be needed to maximize cost- and time-effectiveness. As periodontitis is a multifactorial disease, studies need to be carried out using nonparametric linkage analysis, case–control association analysis and transmission disequilibrium analysis to determine susceptible or resistant genes for periodontitis. Before the endorsement of any such model as a clinical screening tool, the models must be reproducible, biologically plausible and relevant, and of sufficient sensitivity and specificity to warrant application to clinical populations.
References
| [1] | Albandar JM, Rams TE. Global epidemiology of periodontal diseases: an overview. Periodontol 2000 2002; 29: 7-10. |
| [2] | Novak JM. Classification of diseases and conditions affecting the periodontium. In: Newman MG, Takei HH, Klokkevold PR, Carranza FA. Carranza’s Clinical Periodontology. 10th edition. Saunders Company 2006: 100-109. |
| [3] | Page RC, Kornman KS. The pathogenesis of human periodontitis: an introduction. Periodontol 2000 1997; 14: 9-11. |
| [4] | Stabholz A, Soskolne WA, Shapira L. Genetic and environmental risk factors for chronic periodontitis and aggressive periodontitis. Periodontol 2000 2010; 53: 138-53. |
| [5] | Nibali L, Tonetti MS, Ready D, Parkar M, Brett PM, Donos N, et al. Interleukin -6 polymorphisms are associated with pathogenic bacteria in subjects with periodontitis. J Periodontol 2008; 679: 677-83. |
| [6] | Yoshie H, Kobayashi T, Tai H, Galicia JC. The role of genetic polymorphisms in periodontitis. Periodontol 2000 2007; 43:102–132. |
| [7] | Socransky SS, Haffajee AD. The bacterial etiology of destructive periodontal disease: current concepts. J Periodontol 1992; 63: 322-331. |
| [8] | Trott JR, Cross HG. An analysis of the principal reasons for tooth extractions in 1813 patients in Manitoba. Dent Pract Dent Rec 1966; 17: 20-27. |
| [9] | Hirschfeld L, Wasserman B. A long-term survey of tooth loss in 600 treated periodontal patients. J Periodontol 1978; 49: 225-37. |
| [10] | McFall WT Jr. Tooth loss in 100 treated patients with periodontal disease. A long-term study. J Periodontol 1982; 53: 539–549. |
| [11] | Loe H, Theilade E, Jensen SB. Experimental gingivitis in man. J Periodontol 1965; 36: 177-187. |
| [12] | Johnson NW, Griffiths GS, Wilton JMA, et al. Detection of high-risk groups and individuals for periodontal diseases: Evidence for the existence of high-risk groups and individuals and approaches to their detection. J Clin Periodontol 1988; 15: 276–282 |
| [13] | Jenkins WM, Kinane DF. The ‘high risk’ group in periodontitis. J Br Dent 1989; 167: 168-171. |
| [14] | Kinane DF, Hart TC. Genes and gene polymorphisms associated with periodontal disease. Crit Rev Oral Biol Med 2003; 14: 430-449. |
| [15] | Laine ML, Loos BG, Crielaard W. Gene Polymorphisms in Chronic Periodontitis. International Journal of Dentistry 2010. |
| [16] | van der Velden U, Abbas F, Armand S, et al. The effect of sibling relationship on the periodontal condition. J Clin Periodontol 1993; 20: 683. |
| [17] | Corey LA, Nance WE, Hofstede P, et al. Self-reported periodontal disease in a Virginia twin population. J Periodontol 1993; 64: 1205. |
| [18] | Michalowicz BS, Aeppli D, Virag JG, et al. Periodontal findings in adult twins. J Periodontol 1991; 62: 293. |
| [19] | Michalowicz BS, Diehl SR, Gunsolley JC, et al. Evidence of a substantial genetic basis for risk of adult periodontitis. J Periodontol 2000; 71: 1699. |
| [20] | van der Velden U, Abbas F, van Steenbergen et al. Prevalence of periodontal breakdown in adolescents and presence of Actinobacillus actinomycetemcomitans in subjects with attachment loss. J Periodontol 1989; 60: 604-610. |
| [21] | Petit MDA, van Steenbergen TJM, Timmerman MF, et al. Prevalence of periodontitis and suspected periodontal pathogens in families of adult periodontitis patients. J Clin Periodontol 1994; 21: 76–85. |
| [22] | Kornman KS, Crane A, Wang HY, et al. The interleukin-1 genotype as a severity factor in adult periodontal disease. J Clin Periodontol 1997; 24: 72–77. |
| [23] | Karimbux NY, Saraiya VM, Elangovan S, et al. Interleukin-1 Gene Polymorphisms and Chronic Periodontitis in Adult Caucasians: A Systematic Review and Meta-Analysis. J Periodontol 2012 Feb 21. Epub. |
| [24] | Brown LJ, Brunelle JA, Kingman A. Periodontal status in the United States, 1988–91: prevalence, extent, and demographic variation. J Dent Res 1996: 75: 672–683. |
| [25] | Marazita ML, Burmeister JA, Gunsolley JC, et al. Evidence for autosomal dominant inheritance and race-specific heterogeneity in early-onset periodontitis. J Periodontol 1994: 65: 623–630. |
| [26] | Shapira L, Eizenberg S, Sela MN, et al. HLA A9 and B15 are associated with the generalized form, but not the localized form, of early-onset periodontal diseases. J Periodontol 1994; 65: 219–223. |
| [27] | Terasaki PI, Kaslick RS, West TL, et al: Low HL-A2 frequency and periodontitis. Tissue Antigens 1975; 5:286. |
| [28] | Diehl SR, Wang YF, Brooks CN, et al: Linkage disequilibrium of interleukin-1 genetic polymorphisms with early onset periodontitis. J Periodontol 1999; 70: 418. |
| [29] | James JA, Poulton KV, Haworth SE, et al. Polymorphisms of TLR4 but not CD14 are associated with a decreased risk of aggressive periodontitis. J Clin Periodontol 2007; 34: 111-7. |
| [30] | Li S, Yang MH, Zeng CA, et al. Association of vitamin D receptor gene polymorphisms in Chinese patients with generalized aggressive periodontitis. J Periodontal Res 2008; 43: 360-3. |
| [31] | Papillon MM, Lefevre P. Deux cas de keratodermie palmaire et plantaire symetrique familiale (maladie de Meleda) chez le frere et la soeur. Coexistence dans les deux cas d’alterations dentaires graves. Bull Soc Fr Dermatol Syphiligr 1924; 31: 82–87. |
| [32] | Cury VF, Costa JE, Gomez RS, et al. A novel mutation of the cathepsin C gene in Papillon–Lefevre syndrome. J Periodontol 2002; 73: 307–312. |
| [33] | Toomes C, James J, Wood AJ, et al. Loss–of–function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat Genet 1999; 23: 421–424. |
| [34] | Haim S, Munk J. Keratosis palmo-plantaris congenita, with periodontosis, arachnodactyly and a peculiar deformity of the terminal phalanges. Br J Dermatol 1965; 77: 42–54. |
| [35] | Kobayashi T, Sugita N, van der Pol WL, et al. The Fc gamma receptor genotype as a risk factor for generalized early-onset periodontitis in Japanese patients. J Periodontol 2000; 71:1425-1432. |
| [36] | Coletta RD, Graner E. Hereditary gingival fibromatosis – a systematic review. J Periodontol 2006; 77: 753-764. |
| [37] | Hart TC, Zhang Y, Gorry MC, et al. A Mutation in the SOS1 Gene Causes Hereditary Gingival Fibromatosis Type 1. Am J Hum Genet 2002; 70: 943–954. |
| [38] | Seymour RA, Ellis JS, Thomason JM. Risk factors for drug-induced gingival overgrowth. J Clin Periodontol 2000; 27: 217–222. |
| [39] | Cebeci I, Kantarci A, Firatli E, et al. Evaluation of the frequency of HLA determinants in patients with gingival overgrowth induced by cyclosporine-A. J Clin Periodontol 1996; 23: 737-742. |
| [40] | Tamiya G, Shinya M, Imanishi T, et al. Whole genome association study of rheumatoid arthritis using 27 039 microsatellites. Hum Mol Genet 2005; 14: 2305–2321. |