-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Brain and Cognitive Sciences
p-ISSN: 2163-1840 e-ISSN: 2163-1867
2015; 4(2): 15-27
doi:10.5923/j.ijbcs.20150402.01
Candidate Biomarkers and CSF Profiles for Alzheimer’s Disease and CADASIL
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLBowirrat Abdalla1, Bisharat Bishara2, Nseir William3, Omary Muhamad3, Yassin Mustafa4
1Prof. Dr. of Clinical Neuroscience and Population Genetics – Director of Research Center, EMMS Nazareth Hospital, Faculty of Medicine in the Galilee, Bar Ilan University, Israel
2Senior Physician Specialists in Family Medicine, Director of EMMS Nazareth Hospital, Faculty of Medicine in the Galilee, Bar Ilan University, Israel
3EMMS Nazareth Hospital, Faculty of Medicine in the Galilee, Bar Ilan University, Israel
4Rabin Medical Center, Campus Hasharon, Petah Tikva, Israel
Correspondence to: Bowirrat Abdalla, Prof. Dr. of Clinical Neuroscience and Population Genetics – Director of Research Center, EMMS Nazareth Hospital, Faculty of Medicine in the Galilee, Bar Ilan University, Israel.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
The differential diagnosis between Alzheimer’s disease (AD) and vascular dementia (VaD) are still roughly problematic in clinical practice, despite the widely used diagnostic criteria to differentiate between the two disorders. There is an increasing evidence that cerebrovascular dysfunction plays a role not only in vascular causes of cognitive alterations but also in AD. Cognitively patients, with AD, show sometimes mixed degrees of associated vascular lesions in 30-60% of AD cases. In opposition, AD pathology may be present in 40%-80% of VaD patients, thus impeding diagnosis accuracy. Therefore, to eliminate this bewilderment and discrepancies in the diagnosis between the AD and VaD, it is worthy to shed light firstly on a disease that is a microangiopathy and represents VaD with clear milestones and features as is the case of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Studying CADASIL CSF biomarkers profile, will help in the differential diagnosis between both diseases sharing the coexisting neurodegeneration, furthermore, CADASIL is a dominantly inherited mid-adult life disorder causing ischemic strokes, which belongs to vasculopathies and symbolizes a genuine prototype of VaD that provides a valuable opportunity for studying its CSF biomarkers. Secondly, examining and evaluating the CSF biomarkers of AD compared to that of CADASIL. The pathogenesis similarities between CADASIL and early onset AD affecting the small vessels of the brain have suggested plausible molecular mechanisms involved in vascular damage and their impact on brain function and also come from the fact that in both diseases genetic mutations occur. CADASIL mutations in NOTCH3 gene generate toxic protein aggregates (Granular Osmiophilic Material- GOM) in the vicinity of vascular smooth muscle cells (VSMCs) causing degeneration and loss of VSMCs in small arteries and arterioles of white matter regions of the brain that lead to dementia, similar to those attributed to mutant forms of the Amyloid Precursor proteins (APP) and presenilins genes who cause overproduction and accumulations of the toxic Aβ42 protein in the brain and collapse of Aβ42 clearance mechanisms in AD. Despite the presumed pathological similarities, substantial differences between the two phenomena may exist especially in the CSF neurochemical phenotypes. To examine this aspect, which may help in the differential diagnosis, we carried out this review.
Keywords: Biomarkers, CSF, Aß, Tau, Vascular dementia, Alzheimer’s diseases, CADASIL
Cite this paper: Bowirrat Abdalla, Bisharat Bishara, Nseir William, Omary Muhamad, Yassin Mustafa, Candidate Biomarkers and CSF Profiles for Alzheimer’s Disease and CADASIL, International Journal of Brain and Cognitive Sciences, Vol. 4 No. 2, 2015, pp. 15-27. doi: 10.5923/j.ijbcs.20150402.01.
1. Introduction
- Alzheimer's disease (AD) is an insidious neurodegenerative disease and a genetically heterogeneous disorder causing dementia in elderly and leading to a massive burden on AD individuals, their families, and on social and health care systems [1]. Its diagnosis is subjective, definite AD can only be diagnosed after pathological brain specimens are examined by either biopsy or autopsy, and it covers 50-60% of all dementia cases. It is estimated that, by 2050, the number of people aged 80 years or older will approach 370 million worldwide and that 50 percent of those aged 85 years or older will be affected with AD [2]. Causes of the disease are multifactorial; where genetics and environmental risk factors work in harmony to cause the disease [3, 4]. Neuropathological features of AD depends on finding extracellular deposits of β-amyloid peptides (Aβ) that lead to senile plaque formation and intracellular neurofibrillary tangles of hyperphosphorylated tau (p-tau), and total tau protein (t-tau). Which all together represent well accepted biomarkers of AD [5, 6, 7]. However, increasing evidence has suggested that vascular pathology plays a critical role in AD pathogenesis as well [8].
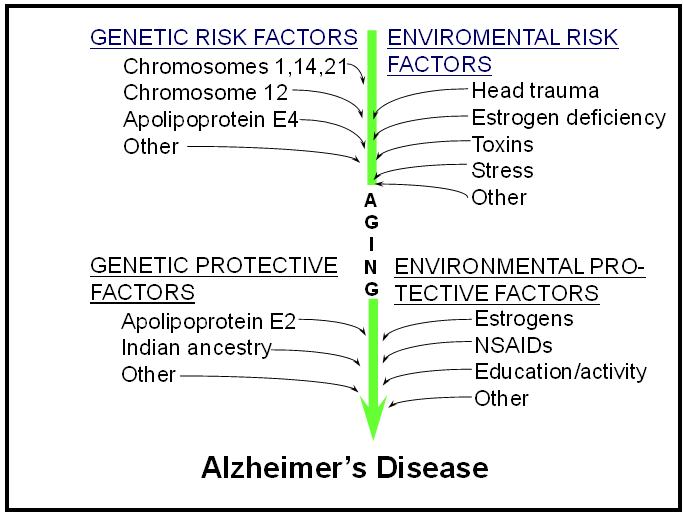 | Risk factors and protective factors for Alzheimer’s disease |
 | The mechanism of Alzheimer’s pathology |
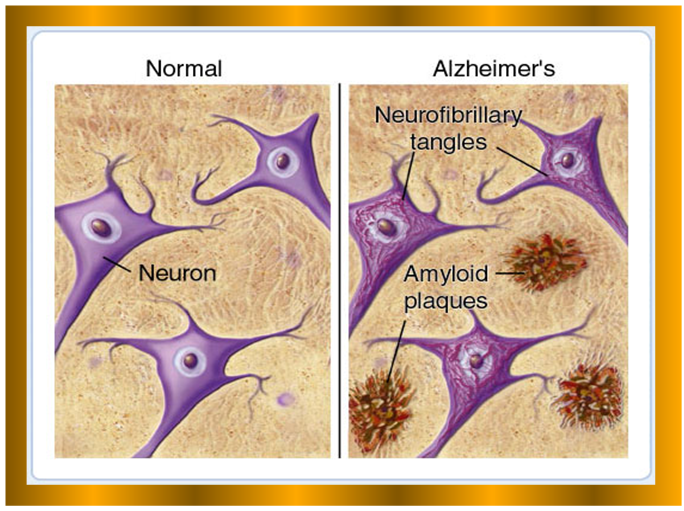 | Comparison between Normal and abnormal neuron cells in AD. |
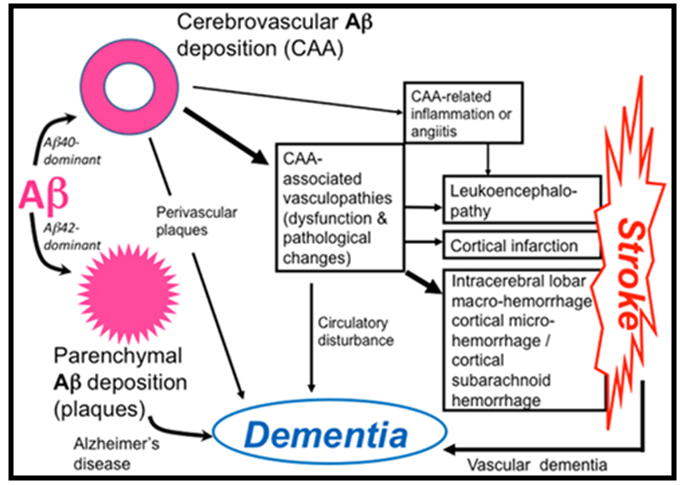 | Pathophysiology of cerebral amyloid angiopathy (CAA)-related disorders. Aβ shows parenchymal or vascular deposition, depending on dominance of Aβ42 or Aβ40, respectively, in elderly individuals and patients with Alzheimer’s disease (AD). Cerebrovascular amyloid deposition, CAA, is related to stroke and dementia. |
2. Overview of Biomarkers Properties
- Access to molecular and biochemical markers of neurodegenerative diseases would complement clinical approaches, and further the goals of early and accurate diagnosis. Hence, the importance of the biological markers studies which are quantitative measurements that provide information about: intrinsic biological processes, a disease circumstances and risk of developing an illness (antecedent biomarkers); assist in diagnosing disease (diagnostic biomarkers); response to treatment (prognostic biomarkers), providing much-needed insight into preclinical and clinical data, all of these are still valid procedures for early detection of diseases [29]. Detection the subject’s susceptibility to the disease prior to appearance of prodromal signs, and detection of neural dysfunction before irreversible cellular damage, will be tremendously valuable for developing; prevention and intervention strategies and early treatments. From here stems the truthfulness and reliability of biomarker to distinguish between normal and interested disease [30]. In fact, biomarkers are set of factors used to measure anatomic, physiologic, biochemical, pharmacological, or molecular parameters associated with the presence and severity of particular disease states or processes in humans and animals [31, 32]. These characteristics are objectively measured and carefully evaluated as indicators of normal biologic or pathogenic processes or pharmacological responses to a therapeutic intervention. Actually, define biomarker panels comprehensively to quantify risk, assess prognosis, and determine response to therapy [33]. Years ago, biomarkers were primarily physiological indicators such as blood pressure or heart rate. More recently, biomarker is becoming an important tool in different disciplines such as in field of oncology, immunology, cardiovascular diseases and metabolic diseases [34]. Synonym for molecular biomarker, such as elevated prostate specific antigen (PSA) as a molecular biomarker for prostate cancer, or using enzyme assays as liver function tests [35]. Biomarkers also cover the use of molecular indicators of environmental exposure in epidemiologic studies such as human papilloma virus (HPV) or certain markers of tobacco exposure such as 4 - (methylnitrosamino) - 1- (3-pyridyl)-1-butanone (NNK) which is a nitrosamine present in tobacco that is a potent procarcinogen. It is activated by CYP2A6, and plays a role as a biomarker of exposure to cigarette smoke, produced upon the curing of tobacco [36, 37]. Also, Genomic biomarkers have principal role in investigation diseases; such as Apolipoproteine Epsilon-4 allele (APOE-ε4) for AD, and HLA for looking for narcolepsy, CD19, Sialophorin, CD11 integrin cluster, and IL-4 receptor – for Crohn’s disease. Other methods and assays are used like; Serum or Spinal fluid substance, Neuroimaging and Physiologic parameters [38, 39, 40, 41]. A. Overview of the CSF Biomarkers for AD and CADASIL.
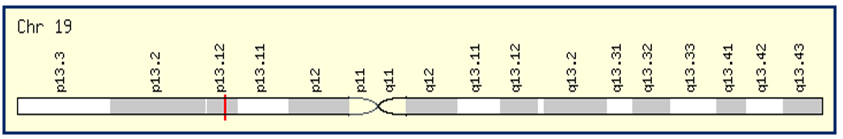 | A. CADASIL Gene located at Chromosome 19p13.2 |
 | B. Amyloid βeta (Aβ) |
 | C. Total tau protein (Microtubule-associated protein tau) |
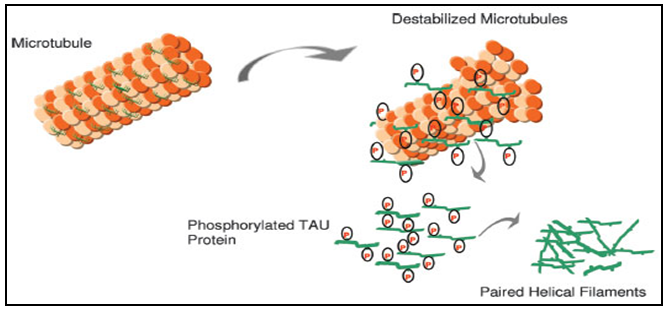 | D. Human Phosphorylated tau (P-tau: phosphorylated at Thr181) |
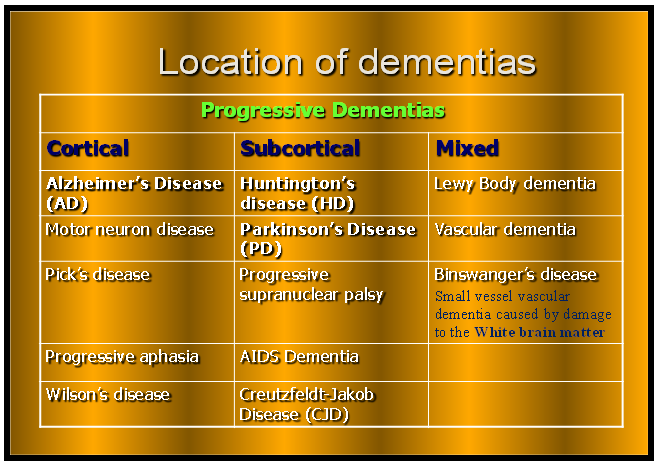 | Different anatomical location of dementias within the brain (Cortical area, Subcortical and Mixed form) |
3. Conclusions
- The differential diagnosis between AD and VaD are still roughly problematic in clinical practice. There is a growing evidence that cerebrovascular dysfunction plays a role not only in vascular causes of cognitive impairments but also in AD. Therefore, to eliminate this bewilderment and discrepancies in the diagnosis between the AD and VaD, it is worthy to examine a disease that is a microangiopathy and represents VaD with clear milestones and features as is the case of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). CADASIL is a dominantly inherited mid-adult life disorder causing ischemic strokes, which belongs to vasculopathies and symbolizes a prototype of VaD that provides a unique opportunity for studying its CSF biomarkers and comparing it with CSF biomarkers of AD. The pathogenesis similarities between CADASIL (a relatively mid-adult disease) with early onset of AD affect the small vessels of the brain suggest plausible molecular mechanisms that are involved in vascular damage and come from the fact that in both diseases genetic mutations occur. CADASIL mutations in NOTCH3 gene generate toxic protein aggregates (Granular Osmiophilic Material- GOM) in the vicinity of vascular smooth muscle cells (VSMCs) causing degeneration and loss of VSMCs in small arteries and arterioles of white matter regions of the brain that lead to dementia, similar to those attributed to mutant forms of the Amyloid Precursor proteins (APP) and presenilins genes who cause overproduction and accumulations of the toxic Aβ42 protein in the brain and collapse of Aβ42 clearance mechanisms in AD. Despite the presumed pathological similarities, substantial differences between the two phenomena may exist especially in the CSF neurochemical phenotypes.
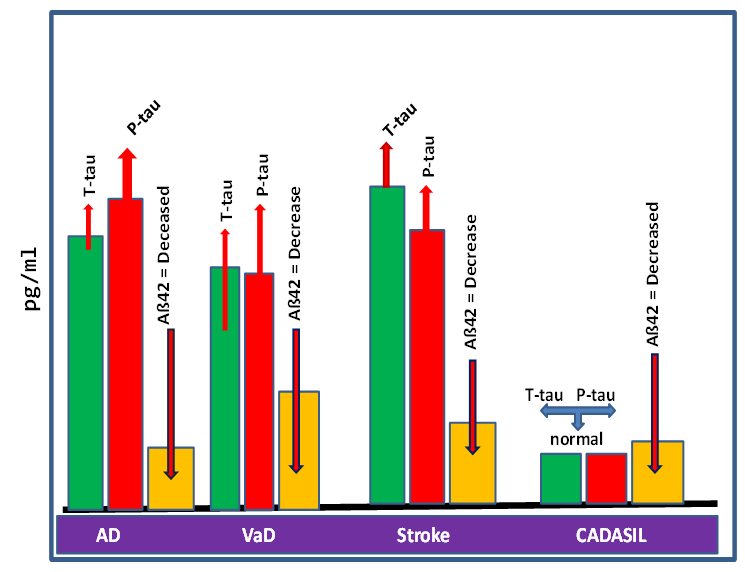 | The figure illustrates the variations in levels of the classical biomarkers Aß42, t-tau and P-tau), altered generally in the AD, VaD, Stroke and CADASIL. |
ACKNOWLEDGMENTS
- We thank Miss Aia Bowirrat for her contribution in revising and editing the manuscript.