-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Journal of Health Science
p-ISSN: 2166-5966 e-ISSN: 2166-5990
2017; 7(3): 44-49
doi:10.5923/j.health.20170703.02

Consanguinity and Autosomal Recessive Mental Retardation in South Waziristan Agency
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLEhsanullah Khan, Jabbar Khan, Muhammad Rafi, Farmanullah Khan, Muhammad Ismail, Attaur Rehman
Department of Biological Sciences, Gomal University Dera Ismail Khan, KPK, Pakistan
Correspondence to: Jabbar Khan, Department of Biological Sciences, Gomal University Dera Ismail Khan, KPK, Pakistan.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Mental Retardation (MR) is characterized by significant limitations in intellectual functioning and adaptive skills occurred before the ages of 18 years. Previously extensive work was done on X-linked MR but now the research work is going on identifying new genes and locus related to autosomal recessive MR. We focused on consanguineous families from South Waziristan Agency (SWA) for linkage analysis of autosomal recessive MR locus. The consanguineous families possessing at least two effected individuals were selected for the study. DNA was extracted by non-organic method and quantitatively measured by NanoDrop spectrophotometer. The DNA of selected families was amplified through touchdown PCR for any possible linkage to already reported MR loci. Linkage analysis was carried out, using microsatellite markers for each locus with forward primer labeled with one of the fluorescent dyes, FAM, VIC, HEX or NED. Linkage was found in family PKMR438 to the locus MRT6, showing that the candidate gene GRIK2 in this region was responsible for MR. Though families PKMR436 and PKMR442 were not linked to known loci but PKMR435 gave a clue of linkage to MRT4 with a marker but, by using genome-wide markers near MRT4 could confirm the linkage. The clinical findings revealed reduced head circumference and deformed hands and feet and difficulties even in sitting in certain patients while no such abnormalities were there in some affected individuals thus showing heterogeneity of autosomal recessive MR genes. We thus assume that more than 50% consanguineous families in this region may show linkage to autosomal recessive MR loci. The X-linked cases may show greater percentage that needs to be explored.
Keywords: Consanguinity, Recessive, Mental retardation, Linkage, PCR, Markers
Cite this paper: Ehsanullah Khan, Jabbar Khan, Muhammad Rafi, Farmanullah Khan, Muhammad Ismail, Attaur Rehman, Consanguinity and Autosomal Recessive Mental Retardation in South Waziristan Agency, Journal of Health Science, Vol. 7 No. 3, 2017, pp. 44-49. doi: 10.5923/j.health.20170703.02.
1. Introduction
- Impairment in growth and development of central nervous system leads to neurodevelopment disorders. When such disorder occurs before 18 years of age characterized by “significant limitations in intellectual functioning and adaptive behaviour”, it is termed as Mental Retardation (MR) while its occurrence after 18 years is called Dementia 1. Such patients face numbers of difficulties in learning, communication, self-care, employment, education and health. Behavioural changes and problems in learning social rules are commonly associated with mentally retarded individuals 2, 3. These signs could easily be found in school going children 4. In autosomal dominant MR, only one copy of the diseased allele is enough to cause disease but is less common condition and very few genes are responsible for such type of inheritance 3, 4. In autosomal recessive MR (ARMR), two copies of a disease allele are required for an individual to be susceptible to express the phenotype. That is why ARMR is observed commonly in consanguineous relationship because the same genetic mutation is transmitted from generation to generation. Consanguinity, which is very common in Pakistan, having socioeconomic, cultural and religious aspects, is the main cause of ARMR. Syndromic ARMR is associated with impairment in vision, nervous system, hearing, skeletal muscles and/or some other impairment. Joubert syndrome, bardet biedl syndrome, Kahrizi syndrome and CAMRQ1 syndrome are the examples of syndromic ARMR. Numbers of genes are responsible for this syndrome including TMEM216, AHI1, NPHP1, CEP290, TMEM67, RPGRIP1L, ARL13B and CC2D2A 5, 6, 7, 8. The children of consanguineous marriages may be at high risk for genetic anomalies because of the expression of autosomal recessive genetic mutations inherited from a common ancestor 7. ARMR is the condition in which the individuals suffer only from cognitive impairment or learning defects and do not have any biological or clinical abnormalities 6, 7. Comparatively, very few genes responsible for ARMR have been reported and are mostly involved in signal transduction cascade 6, 8. The genes are PRSS12, CRBN, CC2D1A, GRIK2, TUSC3, TRAPPC9, TECR, ST3GAL3, MED23, MAN1B1, CRADD, NSUN2 7, 8, 9. There is no proper drug therapy for most of the MR types but certain drugs are used mainly to overcome the complications associated with MR 9. By providing suitable environment, the IQ level of MR individuals can be increased 10.Here we focused on genetic determination of ARMR in families of SWA for the first time through linkage analysis with the objective that this step will become the basis for further characterization in very near future.
2. Materials and Methods
- The families having at least two affected individuals were selected for this study in SWA of KPK, Pakistan. Pedigree of each family was drawn and 5ml blood sample of each individual was collected in EDTA. Detailed information of each family were recorded on studydesigned performa. Total 4 families were selected for further characterization. DNA was extracted by a nonorganic method 11. Five ml of blood was washed with T.E (10 mM Tris HCl pH 8.0, 2 mM EDTA). The pellet was resuspended in 4 ml buffer containing 10 mM Tris HCl of pH 8.3, 2mM EDTA and 450 mM NaCl. 250µg protienase K and 100 µl of 10% SDS were added for digesting protein. It was then put at 65°C for 3 hours. Proteins were precipitated with 0.6 ml of 6M NaCl by shaking it vigorously for 50 seconds and centrifuged at 4000 rpm for 15 minutes. The supernatant was transferred to another 15 ml tube and DNA was precipitated with equal volume of isopropanol 11. After washing with 70% ethanol, DNA was dissolved in 0.5 ml TE and heated at 70°C for 2 hours. DNA was quantitatively measured with NanoDrop spectrophotometer (Thermo Scientific NanoDrop 2000).Preparation of Master Plate and Replica PlatesTo carry out linkage analysis in families affected with RP, a 96-well master plate map was designed in which there was repetition of each sample three times. Both the parents and normal individuals were included in this study alongwith affected individuals.Linkage Analysis and amplificationThe DNA of selected families was amplified through touchdown PCR to see any possible linkage to already reported MR loci Table 1). Linkage analysis was carried out, using microsatellite markers for each locus (Table 1). Forward primer was labeled with one of the fluorescent dyes (FAM, VIC, HEX or NED).
|
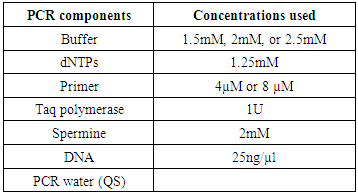
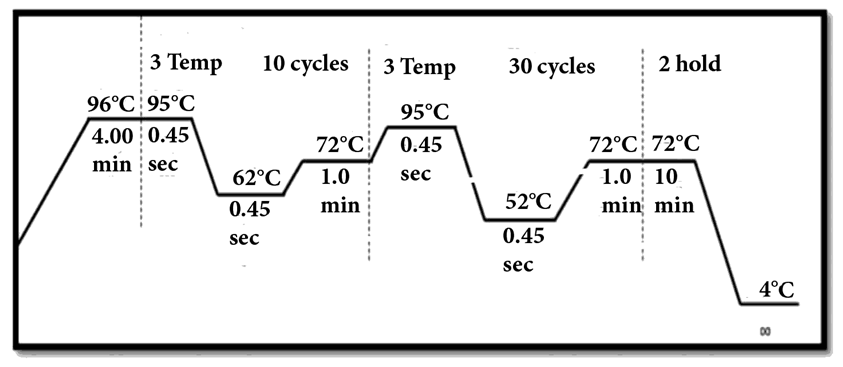 | Figure 1. Conditions for touch down PCR |
3. Results
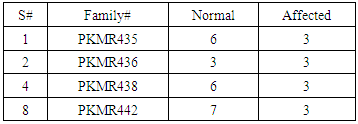
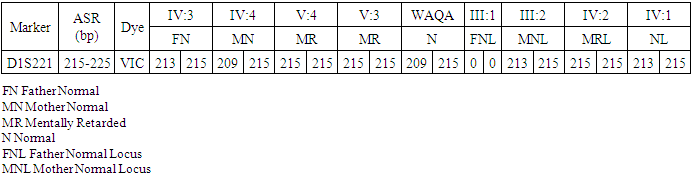
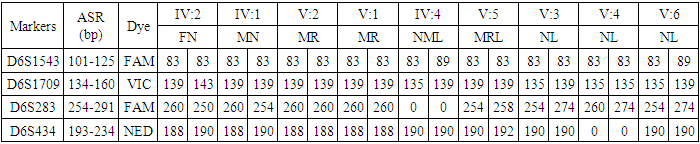
- Four families PKMR435, PKMR436, PKMR438 and PKMR442 (Table 2 and 3) were characterized for linkage analysis.
|
|
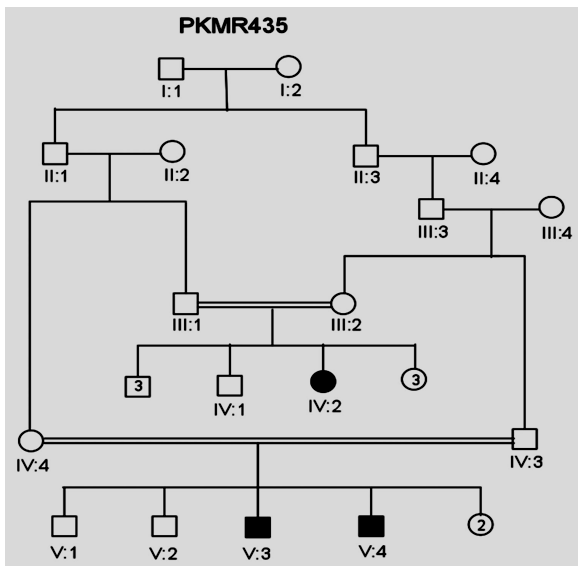 | Figure 2. Pedigree of Family PKMR435. Squares represent males while circles are females. Filled symbols indicate individuals with MR. Double lines between individuals show consanguinity |
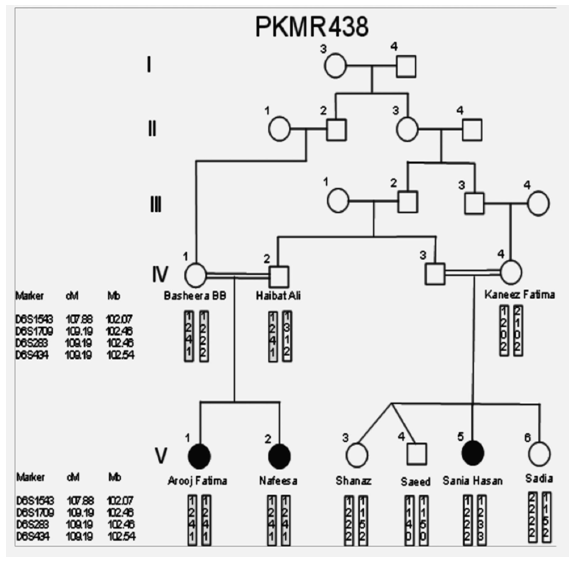 | Figure 3. PKMR 438 Family, squares and circles represent males and females respectively. Filled symbols indicate individuals with MR. Double lines show consanguinity |
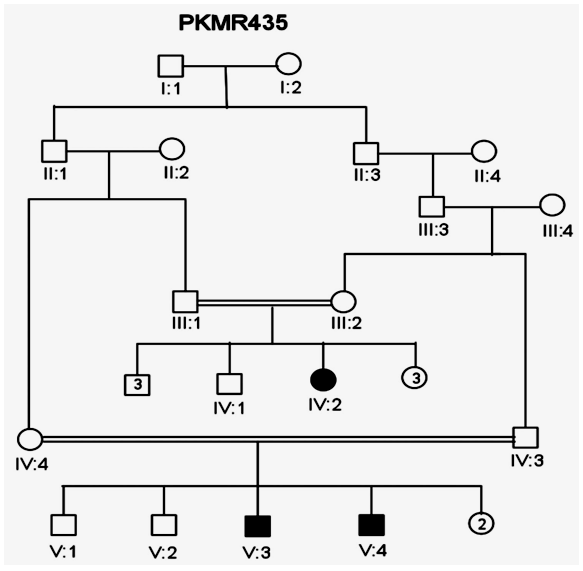 | Figure 4. Family members of PKMR 435. Squares represent males while circles are females. Filled symbols indicate individuals with MR. Double lines show consanguinity |
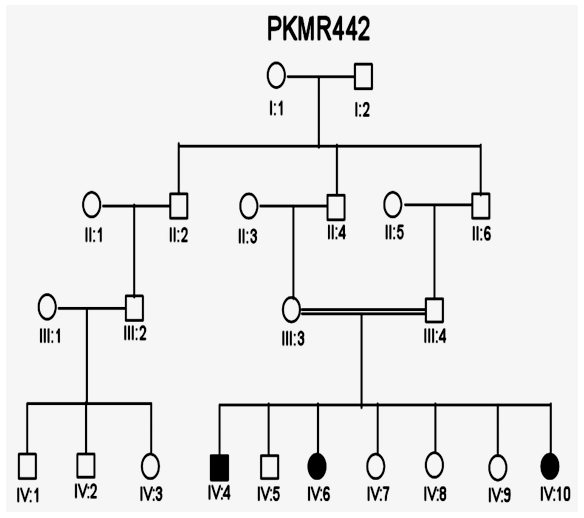 | Figure 5. Pedigree of family PKMR 442 where males and females are shown with squares and circles respectively. Filled symbols are MR patients while double lines indicate consanguinity |
4. Discussion
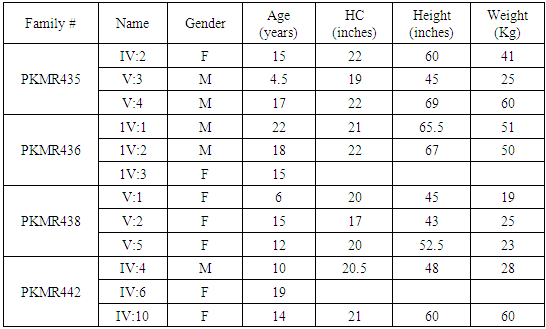
- In the past, little work has been done on ARMR as most of the work has been done on X-linked MR because most of the genes responsible for the mental capacities are located on X chromosome 1. In contrast to X-linked MR, ARMR is very heterogeneous as different mutations in a single gene show variations in phenotypic features 9, 10. The first mutation in GRIK2 gene, found in Iranian family, did not show neurological problems, congenital malformations or facial dysmorphism. The body height, weight, and head circumference of affected individuals were also normal 12. Another mutation in this gene was detected in another affected family and was correlated with wide spectrum of clinical features like delayed development from birth, cognitive impairment and seizures, one individual was also suffering from movement abnormalities like dystonia and tremors 13. The advanced clinical and molecular technologies like next generation sequencing, microarray, chip technology, hybridization, MRI and CT scan are further contributing to the heterogeneity of ARMR. The phenotypic and clinical features of potentially linked family PKMR438 to the locus MRT6 having known MR gene GRIK2 are not completely related to above two cases as the individual V:2 (Table 4) had reduced head circumference, unable to communicate, sit or even crawl Apart from GRIK2, the locus MRT6 also contain 25 annotated genes, of which, seven genes naming CCNC, COQ3, MCHR2, GPR63, KLHL32, POU3F2, and SIM1 are considered to be plausible candidate genes for MR. So further molecular screening needs to decipher this specific family. The countries like Pakistan, where there is high rate of consanguinity there is significant burden on genes responsible for autosomal recessive disorders including MR. Here out of total marriages 62.7% are consanguineous of which 1st cousin marriages are 84% 14. This is called as first degree or close consanguineous union 15. In our study all the 4 enrolled families were with consanguineous union and they were the first cousins of their partners. Though we just followed the criteria of more than one affected individual per family but after pedigree drawing found consanguinity in all the 4 enrolled families. So the data of developed countries where there is equal ratio of genetic and environment factor of MR could deviate from ours due to high rate of consanguinity. Determining genes responsible for MR can help in genetic counselling by avoiding marriages among carriers of the same defective genes 16. In homozygosity mapping the effected siblings should share a common homozygous haplotype transmitted from the heterozygous parents. Once getting genetic affinity of different races through haplotype analysis it will be easier to reconstruct migration history 17. PKMR 438 is Shia family that has strong religious and cultural affiliations with Iran where the MRT6 locus was first identified. The locus MRT4 was first identified in Turkish family 18 and PKMR435 family is tribal Pashtun and is linked to the four markers (D6S1543, D6S1709, D6S283, D6S434) of MRT6. Physical positions of these markers on chromosome 6 are 102073772, 102465322, 102466434 and 102542633 respectively. These markers have genetic distance from 107.88cM to 109.19cM so the distance between linked markers is 1.31cM. GRIK2 is the known gene of this region responsible for MR. Hence this family could be of Turkish origin as Turks invaded this region in medieval ages and settled permanently.
|
|
5. Conclusions
- MRT4 locus is one of the determining factors in MR patients and that the population of SWA needs special attention with respect to genetic counselling, proper screening and awareness about relationship between consanguinity and genetic diseases.