-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Journal of Health Science
p-ISSN: 2166-5966 e-ISSN: 2166-5990
2017; 7(3): 39-43
doi:10.5923/j.health.20170703.01

Haematological and Genetic Characterization of Thalassemia Intermedia in Tank and South Waziristan Agency of Khyber Pakhtun Khwa
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLJabbar Khan 1, Dost Muhammad 2, Zia Ur Rehman 1, Majid Jamal Khan 3, Shahid Niaz 4, Nafees Ahmad 5
1Department of Biological Sciences, Gomal Univeristy, Dera Ismail Khan, Pakistan
2Bannu Medical College, Bannu, Pakistan
3COMSATS Institute of Information Technology Wah Campus, Wah Pakistan
4Department of Zoology, Kohat University of Science & Technology, Kohat, Pakistan
5Institute of Biomedical Sciences & Genetic Engineering, Islamabad, Pakistan
Correspondence to: Jabbar Khan , Department of Biological Sciences, Gomal Univeristy, Dera Ismail Khan, Pakistan.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Thalassemia is an autosomal recessive disorder of hemoglobin synthesis characterized by absence or reduced synthesis of one or the other of globin chains of hemoglobin. This study was designed to address the issue of thalassemia so as to molecularly characterize all the patients for properly managing the course of disease. Blood samples were collected in accordance with criteria for thalassemia intermedia. Hb electrophoresis was done for quantitative measurement of globin chains and polymerase chain reaction to identify both deletions and point mutations in α and β globin genes respectively. A total of 38 thalassemia patients with their ages in the range of 4 to 35 years were characterized. Parents of 32 patients were closely related and only of the 6 were unrelated. Ten β thalassemia mutations; HbS Cd 6 (A>T), Cd41/42 (−TCTT), Cd 8/9 (G), Cd 30 (G>C), IVS I−5(G>C), −88 (C>T), Cap1, HbE, Cd 5 (−CT) and Cd 16 (−C) were identified. HbS Cd 6 (A>T) was found the most prevalent and in all the three ethnic groups of the region but Cd41/42 (−TCTT), Cd 8/9 (G), and −88 (C>T) were found only in Pashtoon ethnic group while IVS I−5(G>C) and Cap1 were found only in Punjabi and Balochi ethnic groups respectively. The α gene rearrangements were found only with patients of thalassemia intermedia. Interestingly −α3.7/−α3.7 genotype was found only with HbS homozygous condition. Moreover, HbS homozygous patients were found for the first time in Balochi ethnic group along−with Pashtoon, while HbE variants were found for the first time in Punjabi patients. Twenty-four patients were identified as having Hb variants. Seven patients were homozygous for HbS and 1 homozygous for HbE while 16 patients were compound heterozygous; 14 for HbS-β- thalassemia and 2 for HbE-β-thalassemia. All thalassemia patients must be molecularly characterized before first transfusion.
Keywords: Autosomal, Hemoglobin, Thalassemia, Ethnic group, PCR
Cite this paper: Jabbar Khan , Dost Muhammad , Zia Ur Rehman , Majid Jamal Khan , Shahid Niaz , Nafees Ahmad , Haematological and Genetic Characterization of Thalassemia Intermedia in Tank and South Waziristan Agency of Khyber Pakhtun Khwa, Journal of Health Science, Vol. 7 No. 3, 2017, pp. 39-43. doi: 10.5923/j.health.20170703.01.
Article Outline
1. Introduction
- Thalassemia is an autosomal recessive genetic disease wherein either of the chains of hemoglobin (Hb) are either not produced at all or are produced at a reduced amount or its variant form is produced [1]. Point mutation, insertion or translocation in the respective gene of glogin chain is the molecular basis of the disease [2]. For its types to classify in, it depends upon which type of chain is either not produced or produced in a reduced amount. Accordingly α, β, γ, δ β, δ, ε, and δβ thalassemia are its main subdivisions [1]. Its geographical distribution varies from region to region. α thalassemia is mainly concentrated in South-East Asians countries while β thalassemia is common in Africa, the Mediterranean region, Indian subcontinent and South East Asia. [3, 4]. In Pakistan β thalassemia is one of the most common inherited Hb disorders 5. Its prevalence is 1.7% [6] to 7.96% [7-9]. Frequency of α thalassemia is estimated by Hb Bart blood in cord blood samples is 2.4% [10]. α thalassemia is characterized by either very little or no production at all of α globin chain of Hb [11] Such a situation may make excessive accumulation of β chains that leads to the formation of γ4 tetramers, β thalassemia is characterized by either no production at all of normal β globin chain, synthesis in a reduced amount or varied forms of β globin chains synthesis. This may lead to accumulation of excessive amount of α globin chains, which may take the shape of insoluble inclusions inside red blood cells (RBCs). These accumulated inclusion ultimately lead to premature destruction of the RBCs while still maturing within the bone marrows and also hemolysis of mature RBC [12]. Thalassemia intermedia patients develop mild to moderate anemia with average Hb level in steady state condition of 7-8g/dl. Such patients are usually associated with mild to moderate jaundice and hepatosplenomegaly. Patients with high Hb levels have no apparent abnormalities in physical development with no typical thalassemic faces. Generally the patients have mild symptoms or are symptom-free, but complications do occur. Iron overload is always manifested with raised plasma ferritin level. Normally patients with β thalassemia intermedia do not require blood transfusion except when they develop infections, which augment anemia. Patients have some what shortened lifespan, but few lead long lives [13-16]. Thalassemia intermedia is a rare condition and very little work has been done in Pakistan. It requires extensive studies to understand the molecular basis and its relation to the phenotype of the patients so as to manage the course of disease and iron overloading. Thus the aim of this research was to determine both α and β thalassemia mutations along with haematological parameters in Tank and South Waziristan Agency, KPK, Pakistan.
2. Materials and Methods
- Blood samples were collected in EDTA. DNA was extracted by a non−organic method [17]. Frozen 5 ml of blood was washed with T.E (10 mM Tris HCl pH 8.0, 2 mM EDTA). The pellet were suspended in 3 ml buffer containing 10 mM Tris HCl of pH 8.0, 2mM EDTA and 400 mM NaCl and then 250µg protienase K was added along with 100 µl of 10% SDS for protein digestion. These were left overnight at 37°C or at 65°C for 3 hours. Proteins were precipitated with 0.5 ml of saturated NaCl by shaking it vigorously for 40 seconds and centrifuged at 3000 rpm for 15 minutes. The supernatant was transferred to another 15 ml tube and centrifuged as above. DNA was precipitated with equal volume of isopropanol [17]. After washing with 70% ethanol, DNA was dissolved in 0.5 ml TE and heated at 70°C for an hour. DNA concentrations were determined by NanoDrop spectrophotometer (Thermo Scientific NanoDrop 2000).Detection of β thalassemia by PCRThe ARMS primers [18] were used to detect β thalassemia mutations. For PCR reaction to perform, 250−300 ng genomic DNA, 240 μM of each dNTP, 1 unit of Taq polymerase, 5.5p mol of each primer i.e. two primers for control fragment and two primers for each of the mutant or control allele and 1x Taq reaction buffer were used in 25 μ reaction volume. The reaction was done through 27 cycles that comprised of one minute denaturation at 94°C, one minute annealing at 65°C and one minute and 30 seconds extension at 72°C. During the first cycle, denaturation was done at 95°C for 5 minutes while the final extension was done at 72°C for 10 minutes. Gel electrophoresis of the PCR product was done on 2% agarose gel that contained ethidium bromide for visualization. Hind III digest was used as a marker.Detection of α thalassemia by PCRThe −α3.7 Kb deletions were detected by amplifying the α globin gene using the forward primer C10: 5/−GATGCACCCACTGGACTTCCT−3/ located in the homologous Y regions of both α1 and α2 genes 19 and the reverse primers C2: 5/−CCATGCTGGCACGTTTCTGA−3/ and C3: 5/−CCATTGTTGGCACATTCCGG−3/, located in the non−homologous 3/ non coding regions of α1 and α2 genes [19]. The PCR was performed in two separate reactions. Thirty μl reaction mix was prepared that consisted of 0.32 μg genomic DNA, 8pmol each of the primers, 220μM each of dNTP, 2mM MgCl2, 12%DMSO, 1 unit of Taq polymerase and 1x Taq reaction buffer [(68mM Tris HCl (pH 8.8), 17mM (NH4)2 SO4, 46μM EDTA, 10mM β mercaptoethanol and 175μg/ml BSA]. The PCR reaction was done through 30 cycles with 1 minute denaturation at 95°C, 1 minute annealing at 52°C and 1 minute and 30 seconds extension at 72°C. Denaturation in the first cycle was done at 95°C for 10 minutes and the final extension was done for 10 minutes at 72°C. Gel electrophoresis of the PCR reaction was done with 2% agarose gel having ethidium bromide for visualization and Hind III digest used as a marker. A normal α1 gene was detected as 2.1 Kb fragment with C10 and C2 primers while −α3.7 mutation was detected with 1.9 Kb fragment. Similarly a normal α2 gene was found with 1.9 Kb fragment with C10 and C3 primers while 2.1 Kb fragment could be found for reciprocal αααanti3.7 if present.
3. Results
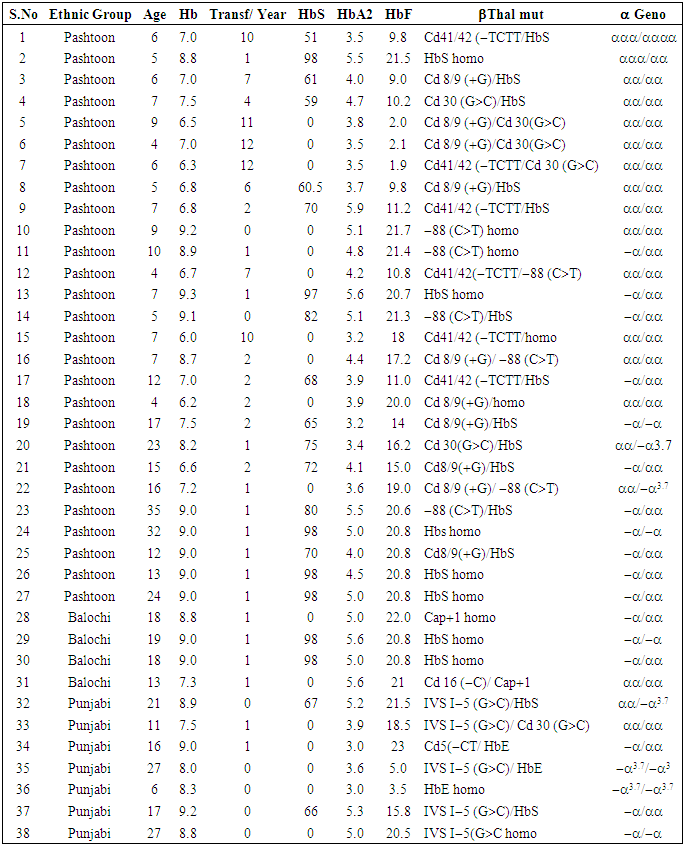
- Thirty-eight clinically diagnosed thalassemia intermedia patients that belonged to three ethnic groups of SWA and FR Tank were analyzed for β thalassemia mutations. Their ages were in the range of 4 to 35 years. Parents of 32 patients were closely related and only of the 6 were unrelated.Screening For The β -thalassemia Mutation The blood samples of 38 patients with 76 β-thalassemia alleles were characterized for β -thalassemia Mutations. Through ARMS techniques, they were screened for 17 known mutation previously reported in Pakistan. Moreover, ARMS technique was also used for the identification of 619 bp deletions. An internal control band of 861 bp was amplified in the all samples to see that PCR reaction was working properly. The normal and mutant primers amplified the normal and the mutant alleles respectively.By using this technique, 76 alleles were characterized for 17 known mutations. Out of 38 β-thalassemia patients, 14 were homozygous and 24 were compound heterozygous (Table 1). Similarly, of the total 38 patients screened out in accordance with criteria for thalassemia intermedia for β -thalassemia Mutations, 5 and 33 were the patients of thalassemia major and of thalassemia intermedia respectively (Table 1). Of the total 14 homozygous patients, 9 belong to Pashtoon ethnic group, 2 Punjabi, and 3 were Balochi. Of the total 24 compound heterozygous patients, 18 were Pashtoon, 5 Punjabi and 1 was Balochi. Ten different β –thalassemia mutations; HbS Cd 6 (A>T) (36.81%), Cd 8/9 (G) (14.48%), –88 (C>T) (11.90%), Cd 41/42 (–TCTT) (9.21%), Cd 30 (G>C) (7.9%), IVS−I−5 (G>C) (7.90%), HbE (5.30%), Cap 1 (A>G) (3.94%), Cd 5 (−CT) (1.31%), and Cd 16 (−C) (1.30%), were identified. HbS Cd 6 (A>T) was the most prevalent mutation (Table 1). There were ethnic differences in the distribution of different mutation. Cd 16 (−C) and HbE were identified for the first time in this region. Interestingly HbE and IVS−I−5 (G>C) were found only in Punjabi while Cd 16 (−C) and Cap1 were found only in Balochi ethnic group. Codon 41/42 ((–TCTT), Cd 8/9 (G) and –88 (C>T) were found only in Pashtoon ethnic group.β Globin chain variantsTwenty-four patients belonging to different ethnic groups were identified as having Hb variants. Seven patients were homozygous and 14 compound heterozygous for HbS−β-thalassemia while 1 patient was homozygous and 2 were compound heterozygous for HbE−β- thalassemia (Table 1).Screening for α- Thalassemia MutationsAll the 38 patients were analyzed for α- thalassemia rearrangements. Analysis of 76 alleles revealed that 16 (42.11%) patients were αα/αα, 13 (34.21%) patients were −α/αα, 4 (10.53%) patients were −α/−α, 3 (7.89%) patients were αα/−α3.7 and 2 (5.26%) patients were −α3.7/−α3.7 (Table 1).
|
4. Discussion
- β Thalassemia is the most common type thalassemia in Pakistan. It is usually diagnosed only clinically by Hb electrophoresis. There is a great need to diagnose thalassemia on molecular basis for proper management of these patients. The most common β thalassemia mutations in Pakistan are IVS-I-5 (G>C) (37.7%), 8/9 (+G) (21.1%), 619 bp del. (12.4%), IVS-I-1 (G>T) (9.5%) and codon 5 (-CT) (3.1%) [8]. The term thalassemia intermedia is clinical definition that is used to describe the patients with phenotypes that are less severe than transfusion dependent thalassemia major but are more severe than the asymptomatic thalassemia trait.Thalassemia is not a rare condition in Pakistan now. During this study HbS Cd 6 (A>T) 13(29.5%) was found to be the most frequent mutations in thalassemia patients, followed by Cd 8/9 (G) (14.48%), –88 (C>T) (11.90%), Cd 41/42 (–TCTT) (9.21%), Cd 30 (G>C) (7.9%), IVS−I−5 (G>C) (7.90%), HbE (5.30%), Cap 1 (A>G) (3.94%), Cd 5 (−CT) (1.31%), and Cd 16 (−C) (1.30%) (Table 1). The clinical severity of β thalassemia depends not only upon the type of β thalassemia mutations but also upon the other factors such as those affecting the α and γ gene expression making these patients either thalassemia intermedia or thalassemia major [14, 16].The age at diagnosis of patients of thalassemia intermedia is in range of 1.5 year to 7 years as compared to patients of thalassemia major having the age at diagnosis less than one year [14, 15]. The present ages of thalassemia intermedia patients were in range of 4 years to 35 years. Unlike thalassemia major, they were not regularly transfused. A small number of patients had 7-10.4 g/dl Hb at the age of diagnosis or just before first transfusion as compared to the patients of thalassemia major, which had hemoglobin level of less than 6 g/dl at the age of diagnosis, or just before first transfusion. The patients of thalassemia intermedia developed few typical symptoms such as splenomegaly, hepatomegaly or skeletal changes usually seen in more severe forms. Only 6 patients had splenectomy. The therapeutic interventions for some of these patients have been folic acid and desferol injections (desfersxamine). Very interestingly, these patients had large amount of HbF in the range of 9%-22% and HbA2 in the range of 3.5% to 5.8% as compared to the patients of thalassemia major, which had less amount of HbF in the range of 1.9-2.2% in the blood taken just before transfusion. We collected blood samples in accordance with criteria for thalassemia intermedia but, after molecular characterization, 5 patients were found as of thalassemia major as their genotypes were β0/β0. Some of the patients of thalassemia intermedia were made transfusion depentent besides the fact that they did not require regular transfusion (Table 1). Hence, in our opinions these values do not indicate the patient’s bone marrow condition because patients get transfusion before their Hb level dropped below 10 g/dl. Therefore these values are a combination of patient’s blood and transfused blood. The parents of most patients were either first cousins or close relatives. If they were not relatives, they belong to same ethnic groups. Interestingly, we found IVS−I−5 (G>C) and HbE in Punjabi ethnic group only, Cd 16 (−C) and Cap 1 (A>G) only in Balochi, and Cd 8/9 (G), –88 (C>T) and Cd 41/42 (–TCTT) in Pashtoon ethnic group only.As previously reported, coinheritance of α thalassemia with β thalassemia reduces the severity of disease [19-23]. In this study we have also found the coinheritance of α thalassemia in the patients of thalassemia intermedia (Table 1). Of the total 38 patients characterized, 16 (42.11%) patients were αα/αα, 13 (34.21%) patients were −α/αα, 4 (10.53%) patients were −α/−α, 3 (7.89%) patients were αα/−α3.7 and 2 (5.26%) patients were −α3.7/−α3.7 (Table 1). Interestingly, the −α3.7/−α3.7 genotype was found only with HbS homozygous condition.Thus all thalassemia patients should be diagnosed molecularly before starting blood transfusion to determine the course of their disease and for their management accordingly.
5. Statistical Analysis
- CONCLUSION: All thalassemic patients should be molecularly diagnosed before transfusion so as to manage the course of disease properly.