-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Frontiers in Science
p-ISSN: 2166-6083 e-ISSN: 2166-6113
2018; 8(2): 27-34
doi:10.5923/j.fs.20180802.01

Modulation of Tumor Necrosis Factor by Raf-1 Inhibitor as a Potential Suppressor of Hepatocellular Carcinoma
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLRehab Bader, Adel A. Guirgis, Hany Khalil
Department of Molecular Biology, Genetic Engineering and Biotechnology Research Institute, University of Sadat City, Egypt
Correspondence to: Hany Khalil, Department of Molecular Biology, Genetic Engineering and Biotechnology Research Institute, University of Sadat City, Egypt.
| Email: |  |
Copyright © 2018 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Hepatocellular carcinoma (HCC) is the most common disorder of liver tumor which often leads to death. Indeed the incidence of HCC has been increased worldwide particularly in developing countries. The serious risk factors associated with HCC development are hepatitis C and B virus infection. Other considered risk factors include alcohol consumption, exposure to environmental toxins such as aflatoxins, hemochromatosis, cirrhosis, diabetes and obesity. A variety of cellular signaling pathways are implicated in HCC such as vascular endothelial growth factor signaling (VEGF), fibroblast growth factor signaling (FGF) and mitogen-activated protein kinase (MAPK). Thus, in the current study we were interested to investigate one of MAPK signals, RAF/MEK/ERK pathway in HCC using HepG2 cells compared with normal hepatocytes cells. The relative gene expression of MAPK and production level of tumor necrosis factor alpha (TNF-α) during cells growth have been `assessed using quantitative real time PCR and quantitative 'sandwich' ELISA assay, respectively. Our results indicate that the growth of HepG2 cells require time-dependent RAF/MEK signaling pathway; the relative expression of RAF and MEK genes are positively associated with cell growth. Further, the production level of TNF-α was increased during HepG2 cells growth in comparison with normal cells. Interestingly, both RAF/MEK pathway and production level of TNF-α are sufficiently regulated in HepG2 cells that were subjected to Raf-1 inhibitor. These data firstly reveal the important clue for exploiting the relative gene expression of RAF/MEK and production level of TNF-α in diagnosis of HCC development. Secondary, our findings provide the potential anti-cancer effect of Raf-1 inhibitor via regulation of RAF/MEK signaling and prevent TNF-α production.
Keywords: Hepatocellular carcinoma, RAF/MEK pathway, TNF-α, Diagnosis and therapy
Cite this paper: Rehab Bader, Adel A. Guirgis, Hany Khalil, Modulation of Tumor Necrosis Factor by Raf-1 Inhibitor as a Potential Suppressor of Hepatocellular Carcinoma, Frontiers in Science, Vol. 8 No. 2, 2018, pp. 27-34. doi: 10.5923/j.fs.20180802.01.
Article Outline
1. Introduction
- Hepatocellular carcinoma (HCC) occurs in the case of chronic liver inflammation that normally raised in males than females with higher incidence in Middle and Western Africa, Southern and Eastern Asia and Micronesia/Polynesia [1]. The major risk factors for HCC include hepatitis B and hepatitis C virus infection, alcoholic and non alcoholic fatty liver [2, 3]. Notably, 50% of HCC cases are associated with HBV infection and almost 25% are associated with HCV [2]. Patient with obesity and type 2 diabetes have been correlated with increased risk of HCC depending on the duration of diabetes and treatment strategy [4]. The initial symptoms of HCC are including yellow eyes and skin, swelling abdomen, varicose veins, weakness, weight loss and fever [2, 5]. Early diagnosis of HCC may facilitate curative treatment; however HCC usually occurred with poor prognosis especially at the early stage. Accordingly, diagnosis of HCC is difficult and often requires more than one imaging to detect the real size of liver tumor so that all treatment options can be offered. Level of alpha-fetoprotein (AFP) and ultrasound are the most common diagnostic tools for HCC in the active surveillance [6, 7]. On the other hand, cell signaling is a type of cellular communications that regulates basic activities and cellular processes. Interruption of cell signaling or cellular processes can initiate various diseases such as autoimmune diseases, diabetes mellitus and cancer. Cell signaling has been widely investigated in human diseases for better understanding of molecular aspect of these diseases, early diagnostic and therapeutical strategy. Several cellular signaling have been identified in HCC such as protein kinase C (PKC) and mitogen activated protein kinases (MAPKs) including RNA activating factor 1 (Raf-1) signaling cascade [8-10]. MAPKs are extracellular communication contain specific proteins chain started from cell receptors to nuclear DNA. MAPK signaling is typically activated through the binding between cell receptors and epidermal growth factors (EGF) so called growth factor pathway or extracellular signal-regulated kinase (ERK). In general, EGF-receptors interaction activates RAS (a small GTPase) followed by stimulation of Raf-1 kinas which subsequently stimulates MEK and transcription factors cMyc to endorse cell division [11]. Contribution of toll-like receptors (TLRs) on macrophages activates MAPKs signaling, which regulates cellular innate immune responses [12]. Deficiency of MAPK signal has been associated with production of pro-inflammatory cytokines such as tumor necrosis factor- alpha (TNF-α) and interleukin -6 (IL-6) and anti-inflammatory cytokine, IL-10, through activation of p38 and c-Jun N-terminal kinase (JNK) [13, 14]. Noteworthy, Raf-1 activating MAPKs signaling has been found to play essential role in evaluation of HCC via activating of cellular machinery processes such as autophagy [15]. Additionally, Raf-1 signaling pathway plays supportive role in viral replications such as hepatitis C virus and influenza A virus [16-18]. In this study, we investigated RAF/MEK/ERK signaling pathway in HepG2 cells compared to normal hepatocytes that propagated in different time points. Raf-1 signaling has been assessed to figure out the possible modulation of TNF-α by Raf-1 inhibitor in propagated HepG2 cells.
2. Materials and Methods
2.1. Cells Lines
- Hepatocellular carcinoma cell line (HepG2 cells) and normal hepatocytes cells that kindly provided from (VACSERA, Giza, Egypt) were propagated in RPMI medium that contains 4 mM L-glutamine, 4 mM sodium pyruvate, 100 U/ml penicillin/streptomycin and 10% bovine calf serum (BCS), and cells were incubated at 37°C and 5% CO2 incubator.
2.2. Raf-1 Inhibitor Treatment
- HepG2 cell line was seeded in 6-well plate in concentration of 200.000 cells per well in 2 ml of RPMI medium and were incubated overnight in CO2 incubator. Then cells were either treated with (100 µg/well) of Raf-1 kinase inhibitor (5-Iodo-3-[(3,5-dibromo-4-hydroxyphenyl)methylene]-2-indolinone, GW 5074, Sigma, USA) or treated with 10 ul DMSO which severed as control.
2.3. Cell Viability and Cytotoxic Effects
- Number of living cells and representative of cell images by inverted microscope have been performed for Raf-1 inhibitor treatment to assess cell viability rate following treatment. Production of lactate dehydrogenase (LDH) in the media from treated cells was monitored in 96-well plate using LDH production kit. According to the manufacture procedures, the same volume of both sample and LDH buffer was incubated for 2 hours followed by 1 hour incubation with LDH substrate then the relative activity of LDH was measured and calculated according to the indicated standard curve. Cell treated with 50 and 100µl of Triton x-100 was used as a positive control [19].
2.4. Total RNA Isolation and cDNA Synthesis
- Cells were collected from cell culture plates in clean and RNase free tubes. Total RNA was isolated from treated and control cells using TriZol (Invitrogen, USA), chloroform methods. Isolated RNA was dissolved in RNase free water and the concentration of all samples was adjusted to final concentration of 100 ng/ul. Then 10 ul from each isolated and purified total RNA was used to generate cDNA using cDNA synthesis kit (Qiagen). According to the manufacturer protocol, total RNA was incubated with reverse transcriptase and poly (dT) primers at 45°C for one hour followed by 5 min incubation at 95°C. The cDNA was then incubated at -20°C until used [20].
2.5. Q-RT-PCR Investigation Parameters
- Q-RT-PCR was used to detect the expression levels of RAF and MEK genes following treatment to investigate the relative expression of RAF-1 and its downstream targets in MAP kinas signaling in HepG2 cells. The resulting cDNA was used as template for subsequent PCR amplification using DNA polymerase and specific primers for indicated genes. The relative gene expression of RAF and MEK were detected using the Quanti-Tect SYBR Green PCR Kit (Qiagen, USA) and oligonucleotides specific for each individual gene Raf-1- sense -5´-TTTCCTGGATCATGTTCCCCT-3´, Raf-1-antisense-5´- ACTTTGGTGCTACAGTGCTCA-3 and Mek-1-For-5´- GACCTGCGTGCTAGAACCTC -3´, Mek-1-Rev-5´- TCTGGACGCTTGTAGCAGAG -3. Levels of GAPDH were amplified using specific oligonucleotides, (sense) 5'-TGGCATTGTGGAAGGGCTCA-3' and (antisense) 5'-TGGATGCAGGGATGATGTTCT-3' which was used for normalization as internal control. The real-time PCR reaction was performed in applied bio-system device with holding temperature of 95°C for 5 min followed by 35 cycles of 15 second duration at 95°C, annealing at 60°C (15 second) and extension at 60°C (45 second). Data analyses of threshold cycle (CT) values were performed using SDS 2.2 [20, 21].
2.6. ELISA
- For quantitative measurement of tumor necrosis factor (TNF-α), sandwich enzyme-linked immunosorbent assay (ELISA) was used to determine serum concentrations of TNF-α using human ELISA kits (Abcam, 181421). Therefore, HepG2 cells and normal cells have been seeded in 96-well plat, and then the cells were either treated with Raf-1 inhibitor or left without treatment. In a time course experiment upon 2, 4, 8, 24 and 48 hours the media were collected and concentration of TNF-α was monitored. Standards and samples were loaded into the 96-wells plate and present TFN-α was bound to the wells by the immobilized antibody. After washing, biotinylated antibody was added followed by HRP-conjugated streptavidin antibody. Finally, a TMB substrate solution was added to the wells and color develops according to the amount of indicated cytokine bound. The intensity of the color was measured at 450 nm [22-24].
2.7. Statistical Analysis
- Microsoft Excel was used to perform final graphs and histograms for our data. Student’s two tailed t-test has been used to investigate the significance of all data that provided by real time PCR analysis. SDS2.2.2 software was used to analyze the qRT-PCT data to drive the Ct values for potential gene expression using delta-delta Ct equations [21, 25].
3. Results
3.1. MAPK Signaling is Activated in Propagated HepG2 Cells
- It has been reported that the extracellular pathway RAS/RAF/MEK, (ERK) plays a key role in the occurrence and development of HCC. To investigate whether activated ERK signaling is detectable in HepG2 cells at RNA levels, the relative gene expression of RAF-1 and its downstream target MEK-1 have been detected in HepG2 cells in-vitro using specific oligonucleotides. HepG2 cells and hepatocytes cells were propagated in 6-well plate with a density of 10x104 cells per well followed by 3 days incubation at 37°C in CO2 (Figure 1A). Total RNA and cDNA have been performed and prepared for qRT-PCR. The analysis of real time PCR thermo cycle’s data showed that the relative gene expression of RAF-1 was increased up to 8 folds in HepG2 cells comparison with normal hepatocytes (Figure 1B). Likely, the relative expression level of MEK-1 was up-regulated more than 4 folds in HepG2 cells compared to normal hepatocytes (Figure 1C). This finding indicates that gene expression levels of RAF-1 and MEK-1 by qRT-PCR reveals the condition of HCC and could be relied as early indicator of the disease.
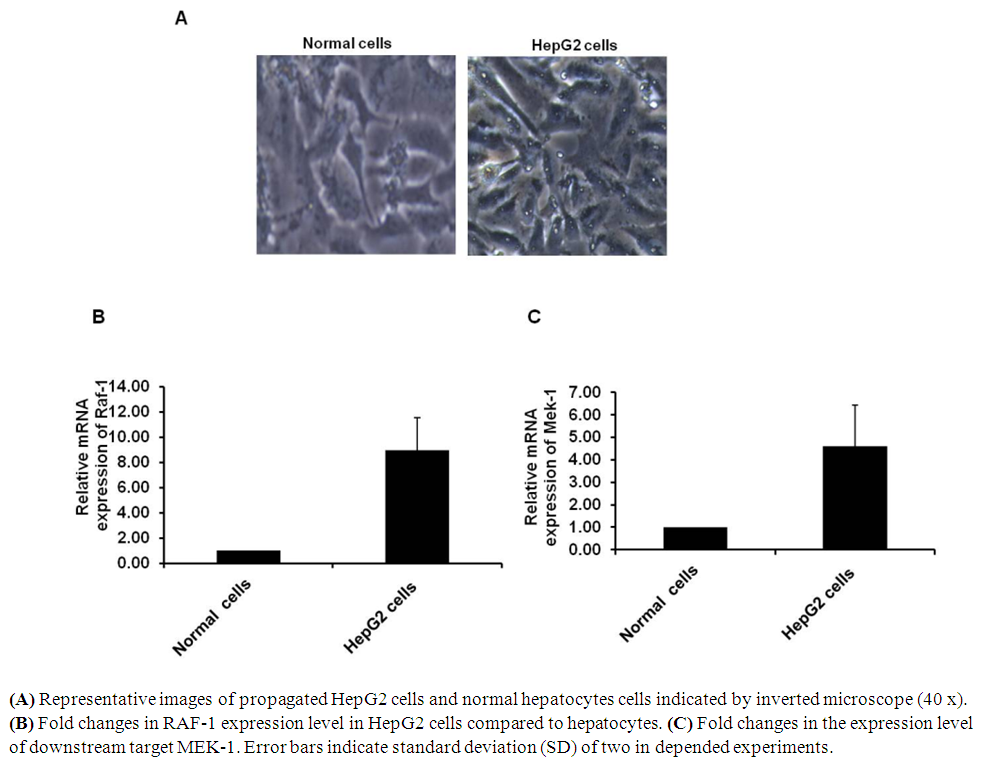 | Figure 1. Activation of MAPKs signaling in HepG2 cells |
3.2. RAF-1 Inhibitor is Secure on Subjected HepG2 Cells
- To investigate the possible and detectable toxic effects of RAF-1 inhibitor on HepG2 cells, number of living cells and cell representative images has been monitored in pre-treated cells. Further, Lactate dehydrogenase (LDH) production from pre-treated cells was assessed in order to investigate cell viability rate of treated cells. LDH is a key enzyme on Krebs’s cycle that activated in living cells and responsible for the conversion of lactate to pyruvic acid during in mitochondria. Secretion of LDH reveals the systematic toxic effect on cell viability rate via stimulating program of cell death (PCD). Thus, HepG2 cells were seeded in 6-well plate with a density of 2 x 105 cells/well followed by overnight incubation. Cells then have been subjected to 100µg/ml of RAF-1 inhibitor followed by overnight incubation. Interestingly, RAF-1 inhibitor showed negligible cytotoxic effects on treated cells indicated by cell images and number of living cells (Fig 2A and B). Moreover, the relative LDH production on treated HepG2 cells showed the same concentration as indicated by untreated cells (Fig 2C). These findings indicate that RAF-1 inhibitor, in concentration of 100µg/ml, is secure to be used as potential mediator of RAF-1 signaling pathway in HepG2 cells.
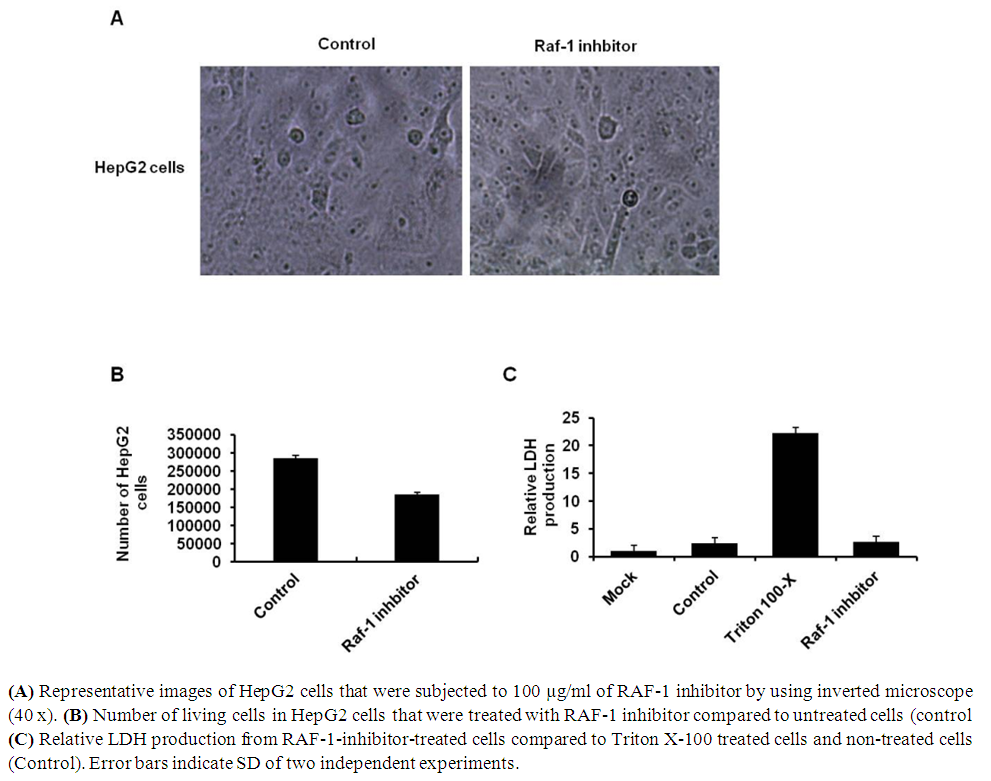 | Figure 2. Cytotoxicity of RAF-1 inhibitor in HepG2 cell lines |
3.3. RAF-1 Inhibitor Efficiently Regulates the Downstream Target of RAF-1 Signaling
- The RAF/MEK/ERK signaling cascades is activated in response to growth factors from their receptors to specific transcription factors which regulates gene expression and prevents apoptosis. Biochemical blocking of RAF-1 activity could regulate cell proliferation and PCD in dose dependent manner. To investigate the effect of RAF-1 inhibitor in the downstream cascades of ERK signaling, HepG2 cells were seeded and were subjected to 100 µg/ml of RAF-1 inhibitor (Invitrogen, USA). The relative gene expression of both RAF-1 and MEK-1 has been detected in overnight-treated cells using qRT-PCR. The result showed that the relative expression of both RAF-1 and MEK-1 were significantly reduced indicated by fold change in treated cells compared to control cells (Figure 3A and B). These data reveal the ability of RAF-1 inhibitor to regulate ERK signaling cascades and the expression of its associated genes in HepG2 cells.
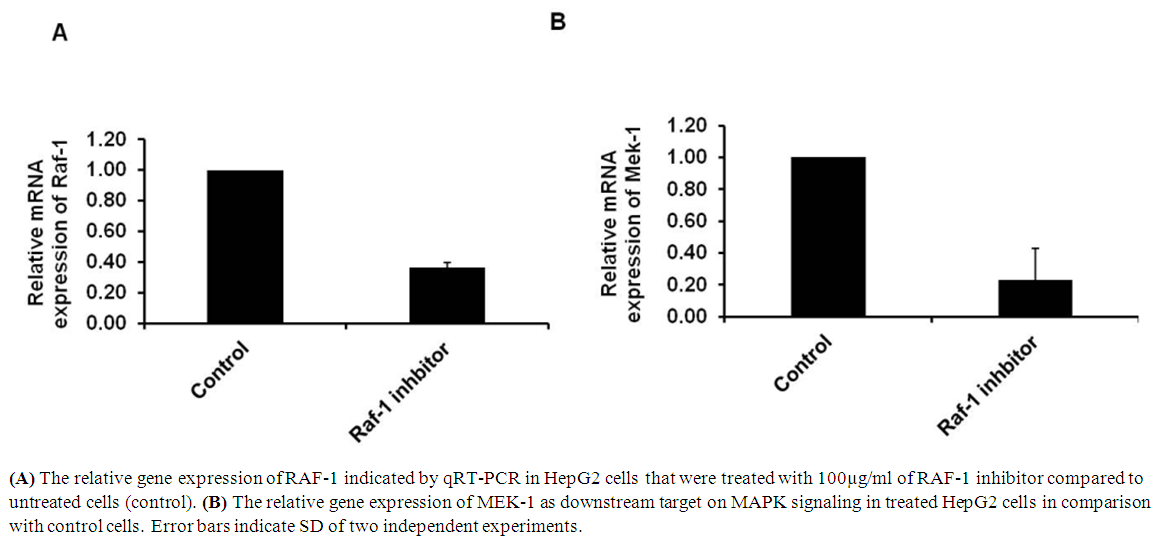 | Figure 3. Regulation of RAF-1 signaling pathway by RAF-1 inhibitor |
3.4. RAF-1 Inhibitor Mediates TNF-α Production in Time and Dose Dependent
- TNF-α is typically produced by hematopoietic cells such as lymphocytes, monocytes, and mast cells. TNF-α is one of the serious pro-inflammatory cytokines which originally characterized by its ability to induce necrosis in tumor cells. To investigate the possible regulation of TNF-α level by RAF-1 inhibitor, HepG2 and normal cells have been subjected to different concentration of RAF-1 inhibitor. Further to test the association of TNF-α level with RAF-1 signaling during cell propagation, the cells were subjected to 100 µg/ml of RAF-1 inhibitor and were propagated for different time points (4h, 8h, 24h and 48h). The concentration of TNF-α has been detected in control and treated cells using ELISA. In the time course experiment, the concentration of TNF-α has been regularly increased in HepG2 cell from 90 pm/ml in 4 hours to 250 pm pm/ml in 48 hours propagation. Meanwhile, the concentration of TNF-α was constant in normal hepatocytes at the different time points (approximately 15 pm/ml) (Figure 4A). In contrast, as a response to RAF-1 inhibitor treatment, the concentration of TNF-α was decreased in treated HepG2 cells in time course dependent from 90 pm/ml in 4 hours to less that 40 pm pm/ml in 48 hours propagation (Figure 4B). Further, production of TNF-α in HepG2 cells that were subjected to different concentration of RAF-1 inhibitor was reduced in dose dependent manner (Figure 4C). These data suggest that the production level of TNF-α has been associated with RAF-1 signaling cascades. Together, a specific inhibitor of RAF-1 regulates gene expression of MEK-1 and blocks the induction of TNF-α in dose and timed course dependent.
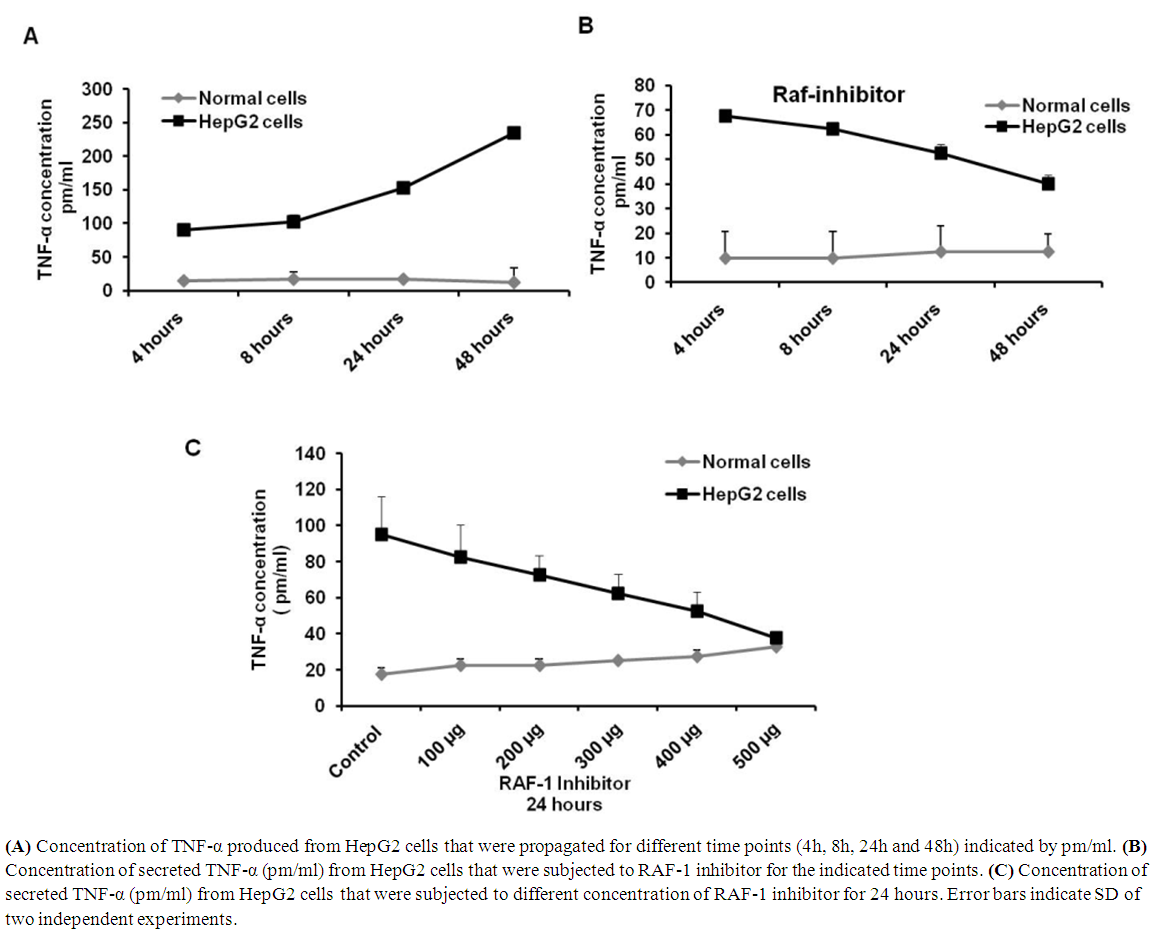 | Figure 4. RAF-1 inhibitor regulates TNF-α in treated HepG2 cells |
4. Discussion
- In the present work we investigated RAF/MEK/ERK pathway in HepG2 cells compared with the normal hepatocytes cells. Our findings showed regular rising of RAF and MEK gene expression in propagated HepG2 cells. Further, production of TNF-α is increased in a time course experiment of HepG2 cells. Interestingly, HepG2 cells that were subjected to RAF-1 inhibitor successfully regulate relative gene expression of MEK and production level of TNF-α in a time and dose dependent manner. Activation of RAF and MEK genes and production level of TNF-α in propagated HepG2 cells elevated by qRT-PCR and ELISA indicate that these techniques can be used to diagnose and evaluate the development of HCC. Additionally, RAF-1 inhibitor has the ability to inhibit MEK gene expression and secretion of TNF-α indicating the possible exploiting of RAF-1 inhibitor as efficacious suppressor for HCC evaluation in-vitro. HepG2 is a kind of cell lines that derived from liver tissue contains a differentiated HCC [26]. HepG2 cells produce a variety of plasma protein such as albumin and fibrinogen in propagated media. Notably, HepG2 cell line is an appropriate system for in vitro investigation of intracellular tracking and cytotoxicity during HCC evaluation and Hepatitis C infection [20, 27]. Therefore, a HepG2 cell line was used to emphasis the possible exploiting of RAF-1 signaling cascades in HCC diagnosis and potential suppressor of cancer development. MAPKs are serine/threonine protein kinases that are typically recognized eukaryotic cells. Four different MAPKs have been previously reported including ERK, c-Jun N-terminal kinase (JNK), ERK5 and p38-MAPK (p38) [28]. ERK cascades contain RAF/MEK/ERK signaling pathway which activated by several stimuli, such as G protein-coupled receptors and receptor tyrosine kinase (RTK) (Figure 5). Activation of ERK signaling regulates gene expression, cell division, differentiation and apoptosis [29, 30]. In cancer cells, several molecular interactions activate ERK signaling such as chromosome ectopic, cytokine mutation and over-expression of mutant receptors such as epidermal growth factor receptor (EGFR) [31, 32]. In HCC, the upstream factor of ERK signal transduction, RAS, has been mutated in 30% of HCC cases while RAF kinase was over-expressed in the most ceases [33]. Additionally many upstream growth factors have been regulated by ERK signaling, such as EGF, vascular endothelial growth factor (VEGF), platelet-derived growth factor-β, (PDGF-β) and transforming growth factor-α (TGF-α) [10, 28]. In apoptosis, RAF/MEK/ERK pathway plays critical role via phosphorylation of various apoptotic regulator, such as caspase-9, Bad, Mcl-1, and the controversial Bcl-2 [28, 34]. Several studies reported the association and direct interfering between the JNK cascade, TNF-α and RAF/MEK/ERK signaling pathway particularly in cancer cells [35-37]. Here, our findings further confirm the regulatory role of RAF/MEK/ERK pathway in production level of TNF-α in HepG2 cells via treatment with RAF-1 inhibitor. Noteworthy, TNF-α is an effective pro-inflammatory cytokine which produced by monocytes, T lymphocytes and cancer cells and plays a critical role in the early events of inflammation [38, 39]. Particularly, TNF-α produced by T-cell plays the major role in autoimmune inflammation and infection-induced super antigen [40]. However, the intracellular signaling pathways that regulate TNF-α in T lymphocytes and cancer cells still poorly understood. Here we show that extracellular signal-regulated kinase (ERK) pathways adjust the transcription and production of TNF-α during HepG2 cells propagation (Figure 5). Therefore, blocking of RAF-1 signaling cascades by RAF-1 inhibitor controls the production of TNF-α and subsequently reduces the inflammatory effects in cancer. Thus, unlike other TNF-α-producing cells, MAPK pathways are critical and cooperate to regulate transcription and production of the TNF-α gene in cancer cells. Accordingly, the present work provides an evidence for the regulatory role of RAF signaling pathway in pro-inflammatory cytokine secretion like TNF-α. Collectively, following of RAF-1 signaling in HepG2 cells and controlling the activation of RAF/MEK/ERK pathway via RAF-1 inhibitor could provide an evidence for crucial role of such a signal in diagnosis and therapy of HCC.
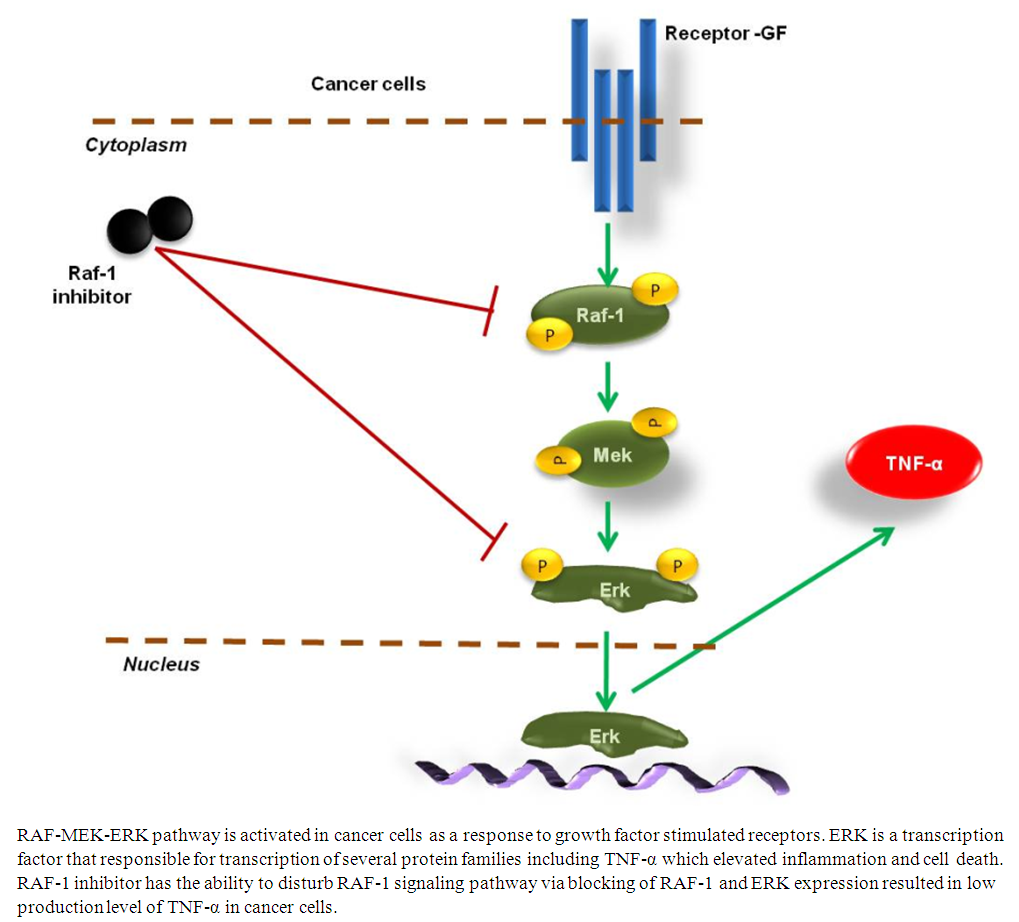 | Figure 5. Schematic representation of RAF-1 signaling pathway and TNF-α production in cancer cells |
ACKNOWLEDGEMENTS
- Working at Dr. Khalil laboratory is supported by Science and Technology Development Fund (STDF), Egypt; through Young Reasecher Program, Project ID: 4694.