-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
International Journal of Diabetes Research
p-ISSN: 2163-1638 e-ISSN: 2163-1646
2016; 5(4): 75-85
doi:10.5923/j.diabetes.20160504.03

Role of Adipokines in Controlling Insulin Signaling Pathways in Type-2 Diabetes and Obesity. Current and Future Perspectives
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLTasneem Bouzid 1, Frederick G. Hamel 2, 3, Jung Yul Lim 1, 4
1Department of Mechanical and Materials Engineering, University of Nebraska-Lincoln, Lincoln, NE, USA
2Departments of Internal Medicine and Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center (UNMC), Omaha, NE, USA
3Research Service (151), Veterans Affairs Nebraska-Western Iowa Health Care System, Omaha, NE, USA
4The Graduate School of Dentistry, Kyung Hee University, Seoul, South Korea
Correspondence to: Jung Yul Lim , Department of Mechanical and Materials Engineering, University of Nebraska-Lincoln, Lincoln, NE, USA.
| Email: |  |
Copyright © 2016 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Adipose cells produce several types of cytokines, known as adipokines, which influence a variety of endocrine signaling pathways. Specifically, several adipokines have been proposed to play a vital role in regulating insulin signaling pathways within various tissues in the body and may thus provide a molecular link between obesity and type 2 diabetes mellitus (type-2 DM). Adiponectin and leptin adipokines have been demonstrated to improve insulin sensitivity, while other adipokines such as resistin, tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) have been evidenced to induce insulin resistance. The low-grade chronic inflammation and greater adipose deposition accompanied by obesity may lead to dysregulation in the production and secretion of adipokines, thus introducing metabolic complications. In this mini review, we will highlight the findings on the role of adipokines in insulin sensitivity/resistance in an effort to better understand the mechanism of type-2 DM. Then, we will provide a perspective on the potential control of adipokine secretion by extracellular mechanical loading cue. This is based on the reported correlation between the whole body exercise and circulating adipokine level. The understanding of cellular level mechanism of the mechanical loading control of adipokine biology may provide a new and improved insight into the missing link among exercise, obesity, and type-2 DM.
Keywords: Adipocytes, Adipokine, Type 2 diabetes, Insulin sensitivity, Insulin resistance, Mechanical loading
Cite this paper: Tasneem Bouzid , Frederick G. Hamel , Jung Yul Lim , Role of Adipokines in Controlling Insulin Signaling Pathways in Type-2 Diabetes and Obesity. Current and Future Perspectives, International Journal of Diabetes Research, Vol. 5 No. 4, 2016, pp. 75-85. doi: 10.5923/j.diabetes.20160504.03.
Article Outline
1. Introduction
- Obesity greatly increases the risk for associated metabolic diseases, including type 2 diabetes mellitus (type-2 DM). Although adipose tissue was thought to serve mainly to store energy in the form of triacylglycerides, numerous studies spanning the last 20 years have provided evidence that adipose also behaves as an endocrine organ by secreting bioactive cytokines known as adipokines. Adipokines have been implicated in mediating insulin signaling pathways in various tissues within the body [1]. The chronic low-grade inflammation and corresponding increase in macrophage infiltration, the characteristics of obese white adipose tissue (WAT) [2], correlate with increased adipocyte secretion of pro-inflammatory molecules such as resistin, tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) [1]. Such molecules impair the ability of tissues to correctly sense and respond to insulin (i.e., causing insulin resistance) via various mechanisms that intervene in glucose transport and lipid metabolism. Conversely, adipocytes are also capable of secreting insulin-sensitizing molecules such as adiponectin and leptin, which improve the response of tissues to insulin and glucose [3].Insulin instigates its metabolic effects by first binding to the insulin receptor (IR) in the plasma membrane, which initiates a phosphorylation cascade. IR is a tetrameric membrane protein consisting of two αβ dimers linked by disulfide bonds and belongs to a family of tyrosine kinases which includes insulin-like growth factor (IGF)-I receptor and the insulin receptor-related receptor (IRR) (see reviews [4-6]). Alternative exon splicing leads to tissue-specific synthesis of two different IR isoforms, IRa and IRb, which have different binding affinities to insulin [5]. IGF-I and IRR are also capable of forming functional complexes with insulin upon binding [4]. Insulin binds to IR at the extracellular α-subunit, which then derepresses the kinase activity of intracellular β-subunit [4-5]. Conformational change and transphosphorylation of specific tyrosine residues on the β-subunit further increases kinase activity, leading to the recruitment of intracellular docking proteins such as insulin-receptor-substrate-1 (IRS-1), -2 (IRS-2), Src-homology/collagen (Shc), and growth factor receptor-bound protein 2 (Grb2) [4-6]. The major substrate for IR in adipose and muscle tissues is IRS-1, which once phosphorylated mediates the association with and activation of phosphatidylinositol-3-kinase (PI3K) [4-7]. Serine phosphorylation caused by cytokines and fatty acid metabolites can inhibit IRS-1 signaling [7]. PI3K can catalyze the formation of phosphatidylinositol4,5biphosphate and phosphatidylinositol3,4,5triphosphate (PIP3), and the latter then recruits protein kinase-B (PKB, or Akt) by binding to its SHT domain [4, 6, 8]. Akt activation results in the translocation of glucose transporters to the membrane, leading to increased glucose uptake by the adipocyte cell. Glucose transporters (GLUT) are facilitative membrane transporter proteins that employ the diffusion gradient of glucose and other sugar molecules across the plasma membrane [9]. GLUT1 is primarily expressed in the brain, with moderate levels of expression in muscle, adipose tissue, and liver [9]. GLUT2, which is important in the sensing of glucose levels, is predominantly expressed in pancreatic β-cells, liver, and kidneys [9]. GLUT3 is largely found in the brain, and displays a strong affinity to for glucose [9]. The insulin-responsive GLUT4 is found in the heart, skeletal muscle, and adipose tissue [9]. The insulin-Akt pathway leads to the translocation of GLUT-4-containing vesicles to the plasma membrane, causing an immediate 10 to 20-fold increase in glucose uptake [9]. Other metabolic downstream effects of the IRS in adipocytes include reduced fatty acid secretion and regulation of the expression of genes important in adipocyte differentiation and lipogenesis [1, 4]. The mitogenic response to insulin is transduced via the mitogen-activated protein kinase (MAPK) cascade, in which Grb2 interacts with IRS and associates with the guanine nucleotide exchange factor son-of-sevenless (SOS) [6, 8]. However, Grb can activate the MAPK pathway independently from IRS by associating with activated Shc [6]. Insulin resistance associated with type-2 DM is a result of various disturbances in the intracellular signaling cascade mentioned above. These include reductions in receptor concentration, kinase activity, IRS phosphorylation, and GLUT transportation [4].Numerous extracellular factors can influence obesity and related adipocyte metabolism and macrophage infiltration. Interestingly, recent research provides increasing evidence of the importance of mechanical loading cue in regulating adipocyte function, particularly in cellular adipogenesis and lipid accumulation and potentially on adipose tissue metabolism and inflammation (see other reviews [10, 11]). In this mini review, we will first briefly highlight the roles that adipokines (adiponectin, leptin, resistin, TNF-α, and IL-6) have been shown to have in regulating insulin sensitivity and resistance. Then, we will explore recent literatures that provide evidence of the mechanical loading control of metabolic regulation in adipocytes. From this review, we expect to suggest a new perspective that extracellular mechanical loading environments of adipocytes may regulate insulin sensitivity/resistance and glucose metabolism via affecting adipokine secretion and its signaling activity.
2. Role of Adipokines in Insulin Signaling
2.1. Adiponectin
- Adiponectin is an adipocyte-specific hormone known to be involved in a variety of metabolic, anti-inflammatory, and vasoprotective activities [12]. Originally termed Acrp30, adiponectin is also referred to as AdipoQ, apM1 (adipose most abundant gene transcript1), or GBP28 (gelatin-binding protein 28) [13]. Adiponectin, the result of apM1 gene, is highly expressed in adipose tissue with nominal expression in muscle and liver. Adiponectin occurs in circulation in the form of three oligomeric complexes for different target tissues and with varying physiological effects, e.g., trimers (low molecular weight, LMW), hexamers (middle molecular weight, MMW), and the oligomeric complexes composed of 18 protomers or more (high molecular weight, HMW) [12]. Adiponectin has four structural domains, N-terminal peptide, variable region, collagenous domain, and globular domain that binds to adiponectin receptors [14]. Upon cleavage by leukocyte elastase, the globular domain is liberated [15]. Then, adiponectin receptors 1 (AdipoR1) and 2 (AdipoR2) serve as receptors for full-length adiponectin and globular adiponectin, though the affinity may vary, e.g., AdipoR1 showing high affinity for globular adiponectin but very low affinity for full-length adiponectin [14].The role of adiponectin in hepatic insulin resistance has been extensively tested, results from which have proposed adiponectin as a potential therapeutic option for patients with type-2 DM [16]. Studies have reported that adiponectin could suppress glucose production and increase insulin sensitivity [13, 14, 16-19]. As in Figure 1, adipocytes manipulated to overexpress adiponectin or incubated in adiponectin-conditioned media displayed increased insulin-stimulated glucose uptake [18]. In unfed mice, adiponectin could sensitize hepatic reactivity to insulin by reducing hepatic glucose production and thus serum glucose levels [13]. The insulin-sensitizing effect of thiazolidinedione (TZD), one of the anti-diabetic agents that promotes glucose homeostasis without increasing insulin release, was diminished in diabetic obese mice by a targeted mutation in the adiponectin gene, thus indicating that adiponectin may play a vital role in peroxisome proliferator-activated receptor γ (PPARγ)-mediated insulin sensitization [3]. Ligand-activated PPARγ heterodimerizes with the retinoid X receptor and regulates transcription by binding to specific PPARγ-responsive elements within the promoters of target genes via regulatory mechanisms that involve 5’-AMP-activated protein kinase (AMPK). This process is thought to eventually result in increased fatty acid oxidation, which may in turn divert the flux of free fatty acids from the muscle and liver to the adipose tissue [20, 21].
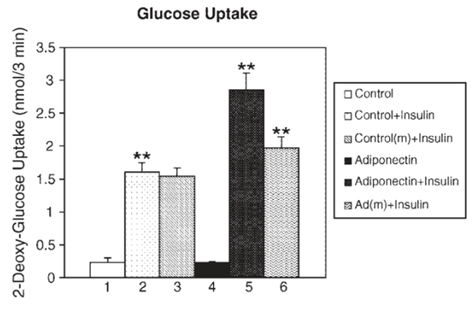 | Figure 1. Mature adipocytes overexpressing adiponectin or incubated in adiponectin-conditioned medium (m) enhances their rate of glucose uptake in response to insulin. Means ± SEM from three separate experiments are shown. **: p < 0.01 for Adiponectin+Insulin vs. Control+Insulin and for Ad(m)+Insulin vs. Control(m)+Insulin. Reprinted with permission from American Society for Biochemistry and Molecular Biology [18] |
2.2. Leptin
- The 16 kDa obese (ob) gene product, leptin protein, was discovered to be a critical regulator of body weight. Named from the Greek root leptόs meaning thin, leptin increased energy expenditure and reduced food intake in obese mice resulting in a 30% reduction in body weight over 2 weeks [26]. Leptin secreted from adipocytes have been suggested to have an important role in the regulation of a variety of body functions such as appetite, satiety, food intake, reproductive function, fertility, puberty, activity, energy expenditure, and atherogenesis (see the review [27]). It was shown that the level of circulating leptin increased under increased caloric intake, and leptin resistance was produced after overfeeding [28]. The infusion of recombinant leptin in voluntarily overfed rats did not alter the contribution of gluconeogenesis to total glucose output, whereas the infusion nearly doubled it in control and pair-fed rats (Figure 2) [28]. Among obese humans, the leptin level strongly correlated with body mass index (BMI) [29]. Moreover, the correlation between leptin and insulin has been relatively well established to be dependent on obesity [30]. On the other hand, some studies reported a correlation less dependent on measures of obesity. For instance, it was demonstrated for lean rats that hyperinsulinemia can upregulate obese gene expression and hypoinsulinemia can downregulate it; while in obese rats, obese gene expression can only be upregulated by hyperinsulinemia [31]. Additionally, treating obese mice with leptin improved overall glucose clearance even prior to the weight loss [32].
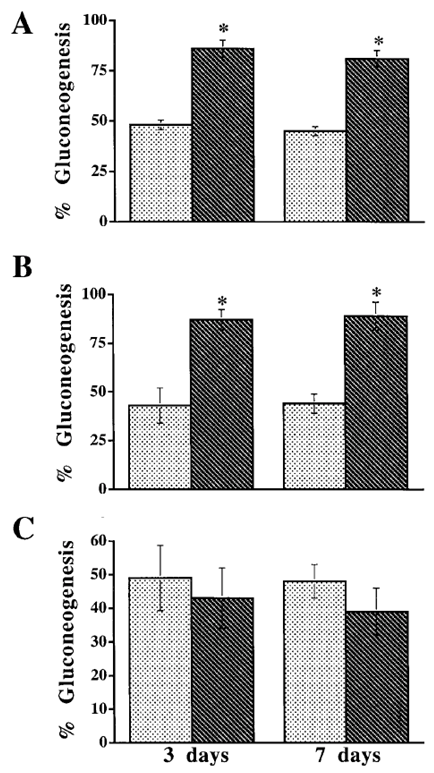 | Figure 2. In control (A) and pair-fed (B) rats, moderate increases in circulating leptin levels caused a marked increase in gluconeogenesis relative to total glucose output. In overfed rats (C), increased leptin levels failed to change the contribution of gluconeogenesis to the total glucose output.  , Lep-; , Lep-;  , Lep+. Means ± SEM. *: p < 0.01 between Lep-and Lep+. Reprinted with permission from American Diabetes Association [28] , Lep+. Means ± SEM. *: p < 0.01 between Lep-and Lep+. Reprinted with permission from American Diabetes Association [28] |
2.3. Resistin
- Resistin is a member of cysteine-rich proteins known as found-in-inflammatory zone (FIZZ) or resistin-like molecules (RELM) which include RELM-α (FIZZ-1), RELM-β (FIZZ-2), and RELM-γ (FIZZ-3). Named for its ability to induce insulin resistance, resistin was discovered to be circulating in mouse serum with increased levels in both genetically- and diet-induced obese mice [39]. Interestingly, resistin has become a focus of several recent studies due to its potential to provide an insight into the possible mechanisms involved in insulin signaling [40]. In mice, resistin is secreted from adipose tissue and acts on skeletal muscle myocytes, hepatocytes, and adipocytes. Exposure to higher glucose concentrations significantly increased resistin expression in 3T3-L1 adipocytes, while treatment with insulin reduced it [41]. Resistin was found to adjust flux through G6Pase by regulating AMPK phosphorylation in the liver, indicating a possible overlap in the mechanism of adipose-derived hormones such as adiponectin (described above) [14, 40]. For example, resistin caused a decrease in AMPK phosphorylation and thus the rate of gluconeogenesis in mice [40]. Moreover, significant decreases in the phosphorylation of Akt and glycogen synthase kinase3 (GSK3), key effectors of the insulin signaling pathway, in human liver cells were induced with resistin in a dose-dependent manner [42].Although the insulin desensitizing effects of resistin in rodents is well documented, studies investigating the link between resistin and diabetes in humans are conflicting. Both clinically and experimentally, researchers found little or no relationship between resistin and insulin resistance. Resistin was expressed in the adipose tissue from obese patients [43], while other researchers showed that isolated adipocytes express very low resistin mRNA levels [44, 45]. The inconsistency may be due to the differences in the genomic organizations of mouse and human resistin in that human resistin is only 59% identical to its mouse counterpart at the amino acid level [46]. On the other hand, resistin was revealed to be relatively more abundant in peripheral mononuclear cells [45]. Thus, the higher levels of resistin found in the adipose tissue may originate from a greater proportion of mononuclear cells obtained from patients with severe obesity rather from the adipose cell itself [1, 2, 45]. Together, resistin affects insulin sensitivity by regulating the activity of Akt and GSK3, key intermediates of the insulin signaling. However, results from both in vivo and in vitro human studies showed inconsistency, potentially due to differences in the genetic sequence code. More studies to clarify such contradiction would be helpful for a potential clinical application of resistin.
2.4. TNF-α
- In 1975, it was discovered that endotoxins induced the host to release a substance, which was called tumor necrosis factor, TNF [47]. TNF-α-converting enzyme (TACE) cleaves the 26-kDa precursor into the secreted 17-kDa form under exposure to stimuli that triggers inflammatory reaction such as oxidative stress [48]. In addition to inflammatory cells such as macrophages and lymphocytes, non-inflammatory cells including adipocytes have also been evidenced to express TNF-α mRNA [49]. Thus, as a primary pro-inflammatory cytokine, TNF-α has been of interest for its possible effects on adipogenesis, lipid metabolism, and insulin sensitivity [50, 51]. Numerous studies have established for TNF-α a role to induce insulin resistance in both animals and humans [49-57]. Endogenous adipose expression of TNF-α was observed higher in obese humans in comparison with controls [49, 52], having a positive correlation with BMI [58]. In a study that transplanted TNF-α-deficient (KO) mice maintained on a high-fat diet with either TNF-α-sufficient (TNF-α+/+) or TNF-α-deficient (TNF-α-/-) bone marrow, KO TNF-α-/- mice exhibited increased insulin sensitivity (Figure 3) as well as decreased PPARγ mRNA levels [51]. Recently, patients with non-diabetic rheumatoid arthritis showed improved insulin sensitivity in response to anti-TNF-α therapy [53]. Moreover, TNF-α receptor (TNFR)-immunoglobulin G (IgG) treated mice required substantially more glucose to maintain normal glucose level [49], and the administration of TNF-α reversed the insulin effects on glycemic levels [54].
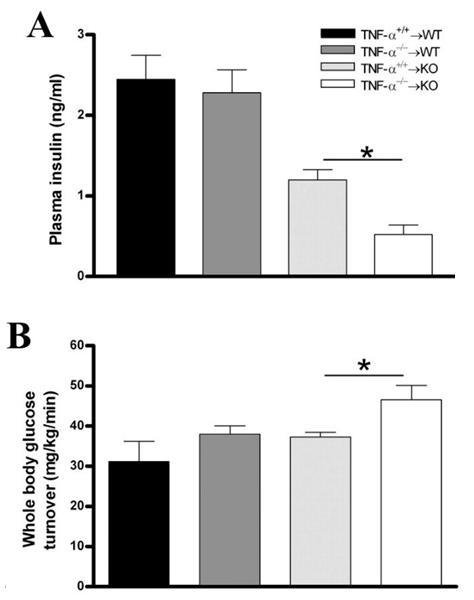 | Figure 3. Wild-type  and knock-out and knock-out  mice underwent bone marrow transplantation and received either mice underwent bone marrow transplantation and received either  or or  bone marrow from donor mice. The mice were put on a high-fat diet (HFD) for 26 weeks. Measured plasma insulin level (*: p = 0.002; A) and whole body glucose turnover (*: p = 0.02; B) indicate that mice lacking TNF-α had the highest insulin sensitivity. Reprinted with permission from the American Physiological Society [51] bone marrow from donor mice. The mice were put on a high-fat diet (HFD) for 26 weeks. Measured plasma insulin level (*: p = 0.002; A) and whole body glucose turnover (*: p = 0.02; B) indicate that mice lacking TNF-α had the highest insulin sensitivity. Reprinted with permission from the American Physiological Society [51] |
2.5. IL-6
- IL-6 is a multifunctional cytokine that plays numerous roles in addition to controlling immune cell function, such as behaving as a hepatocyte-stimulating factor and a growth factor for human myeloma and promoting the transition of hematopoietic stem cells from the G0 to G1 phase [59]. The binding of IL-6 to its low affinity receptor, IL-6 receptor-α (IL-6Rα), forms a high affinity hexameric complex with two gp130 proteins to activate the JAK/Tyk tyrosine kinase and STAT pathways [60]. IL-6Rα is abundantly detected on the surface of certain cells including macrophages, neutrophils, myocytes, and hepatocytes [61].There has been an interest in clarifying the potential role of IL-6 in type-2 DM and insulin sensitivity. Although patients with type-2 DM have shown elevated circulating IL-6 [62], results from studies examining the correlation between IL-6 and insulin resistance have not been fully consistent. IL-6 has been shown to activate SOCS-1 and -3 proteins in the liver, thus accompanying insulin resistance [63, 64]. However, another study reported that while IL-6 increased SOCS-3 expression in murine myotubes, glucose uptake was increased rather than decreased [65]. It was shown that prolonged IL-6 exposure caused insulin resistance in both differentiating and fully differentiated adipocytes accompanied by decreased expression of PPARγ [66]. As in Figure 4, the treatment of differentiating preadipocytes with IL-6 significantly reduced both lipogenesis and glucose transport. Moreover, IL-6 could also downregulate the expression of adiponectin, the insulin signaling sensitizer as described above, in 3T3-L1 adipocytes [67]. On the other hand, a recent study showed that global deletion of IL-6 in mice induced maturity-onset obesity, liver inflammation, and whole-body insulin resistance [68].
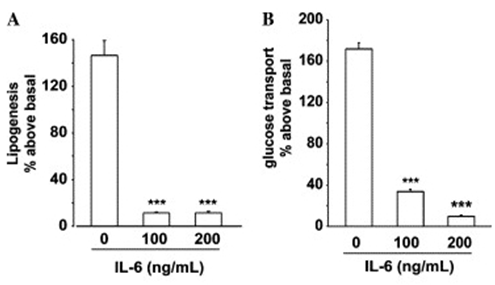 | Figure 4. Administration of IL-6 (100 and 200 ng/mL) during differentiation of 3T3-F442A preadipocytes greatly reduces (A) lipogenesis and (B) glucose transport at maturity. Each bar represents mean ± SEM of 3-4 experiments performed in duplicate. All values are expressed as percentages above basal (control) measurements. ***: p < 0.001. Reprinted with permission from Elsevier [66] |
3. Perspective on the Potential Role of Mechanical Environment in Adipokine Secretion and Insulin Sensitivity
- Physical exercise has been suggested as a powerful tool not only to control body weight but also to improve systemic insulin sensitivity. The anabolic control of insulin signaling activity through dynamic physical exercise can be achieved through enhancing glucose transport, regulating hepatic glucose output, and repairing lipid metabolism. Studies have tested the effects of human exercise on the expression of adipokines (see detailed reviews in [71, 72]). These include testing of both moderate aerobic exercise and resistance training protocols incorporating cycling, running, and ramp exercises [73-75]. These studies have reported that regular exercise training, sometimes even after a single bout of exercise, may cause a reduction of pro-inflammatory adipokine expression while inducing insulin sensitivity supporting adipokine secretion. Obese young women, after the exercise program, showed significantly increased level of circulating adiponectin while displaying decreased circulating TNF-α (Figure 5) [73]. On the other hand, the level of leptin, another insulin sensitizing adipokine, decreased after exercise. While it is not fully established, it may be hypothesized that mechanical loading applied to the adipose tissue during exercise may play a role in the exercise control of insulin sensitivity/resistance via affecting the level of insulin signaling regulatory adipokine secretion.
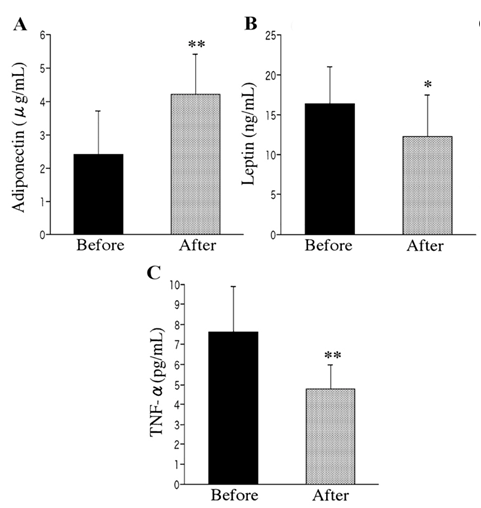 | Figure 5. Levels of circulating adiponectin (A), leptin (B), and TNF-α (C) in obese young women before and after a single bout of moderate intensity exercise. The results indicate that the exercise increases plasma adiponectin while decreasing plasma leptin and TNF-α. Each bar represents mean ± SEM. *: p < 0.05 and **: p < 0.01. Reprinted with permission from the Japan Endocrine Society [73] |
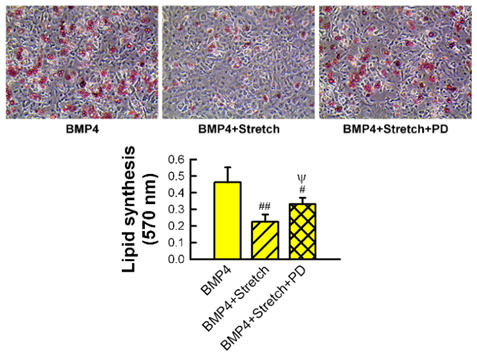 | Figure 6. Bone morphogenetic protein 4 (BMP4)-induced lipid synthesis in murine MSCs during adipogenic differentiation is impeded by cyclic stretching. The inhibitory effect of cyclic stretch on adipocytic differentiation is reversed by the addition of ERK inhibitor, PD98059 (PD). Oil red O stained images and lipid quantification by spectrophotometer. Comparison with BMP4 (#, ##) and BMP4 plus stretch (ψ) shown at p < 0.05 (#, ψ) and p < 0.01 (##). Reprinted with permission from Elsevier [85] |
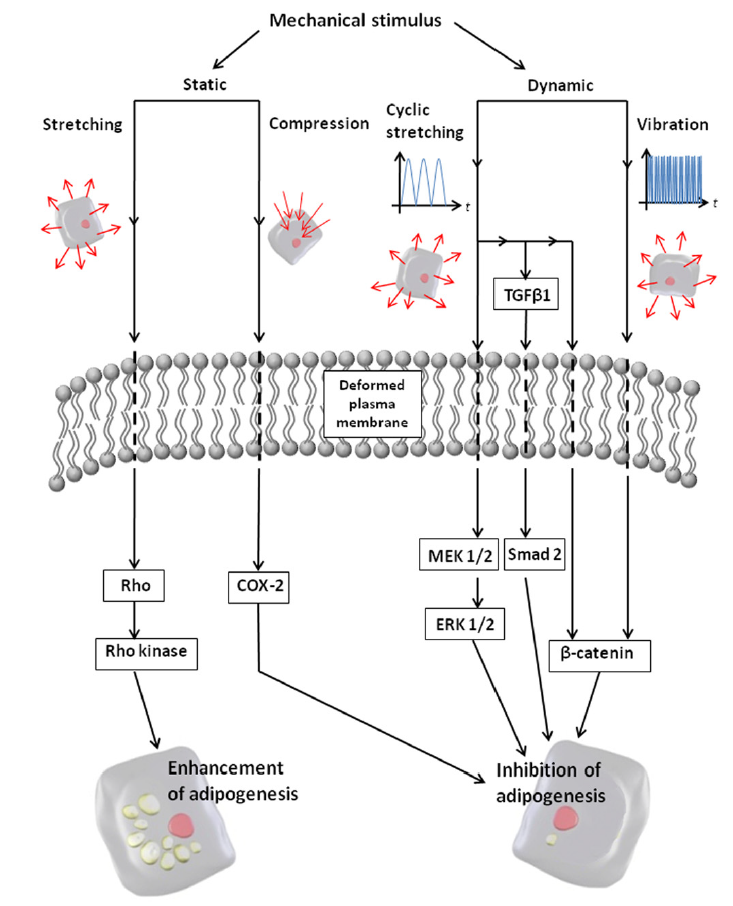 | Figure 7. Potential mechanotransduction pathways involved in cellular adipogenesis. Static stretching activates the Rho and Rho Kinase (ROCK) signaling pathway, leading to enhanced adipogenesis. Cyclic stretching activates ERK, TGFβ1/Smad, and β-catenin pathways, leading to reduced adipogenesis. Reprinted with permission from Elsevier [76] |
4. Conclusions
- Researches provide strong evidence that adipose tissue plays important roles outside of energy storage. Adipose tissue has been shown to behave as an endocrine organ capable of regulating many physiological processes by the secretion of protein factors known as adipokines. Of great clinical relevance to type-2 DM in particular are adiponectin, leptin, resistin, TNF-α, and IL-6. Adiponectin has long been of great interest to researchers as an insulin-sensitizer and a potential target of anti-diabetic drugs, and leptin is a multifunctional protein that has been shown to reverse insulin resistance. Resistin, on the other hand, has been shown to target key nodes of the insulin signaling pathway to induce insulin desensitization. TNF-α is a pro-inflammatory cytokine thought to be increased in obese individuals due to the chronic low-grade inflammation associated with obesity, and has been shown to also induce insulin resistance. While further research is still required, IL-6, a multifunctional cytokine, has been shown to alter insulin sensitivity both in vivo and in vitro studies. Recent advances in research indicate that physical exercise may reduce pro-inflammatory adipokine expression while improving insulin sensitivity by causing an increase in the secretion of insulin regulatory adipokines. Furthermore, research is providing increasing evidence that adipose tissue is mechanically responsive, much like muscle and bone. Thus, it may be of interest to investigate the cellular level effect that mechanical loading environment has on the adipokine secretory response of adipocytes in an effort to further illuminate the role of adipokines in insulin signaling. The broader implications of such research include a more advanced understanding of type-2 DM and revealing potential novel targets for anti-diabetic drugs.
ACKNOWLEDGEMENTS
- This literature review was supported by NSF Graduate Research Fellowship 1610400 (awarded to Bouzid), Nebraska Tobacco Biomedical Seed Grant (awarded to Hamel and Lim), NSF CAREER Award 1351570, Nebraska Research Initiative, and UNL Layman New Directions Awards (all awarded to Lim). We also thank the support from the Department of Veterans Affairs (for Hamel).