-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Clinical Practice
p-ISSN: 2326-1463 e-ISSN: 2326-1471
2015; 4(1): 1-5
doi:10.5923/j.cp.20150401.01
Non ∆F 508 Cystic Fibrosis Diagnosed at Age 50 in an African American Male: A Primary Care Perspective
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLAnthony Otekeiwebia, Adesorji Oderinde, Chinedu Ivonye
Morehouse School of medicine, Atlanta, Georgia, United States
Correspondence to: Anthony Otekeiwebia, Morehouse School of medicine, Atlanta, Georgia, United States.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
We present a case of a 50 year old African American male who presented with a productive cough and found to have bronchiectatic changes on his imaging studies, a sweat test was positive and subsequently a genetic analysis was done which showed a 711 mutation with deletion in intron 5. Identification of patients with atypical cystic fibrosis is of particular importance to the primary care physicians because appropriate referral and treatment can substantially affect quality of life and life expectancy.
Keywords: Atypical cystic fibrosis, Adult, African Americans
Cite this paper: Anthony Otekeiwebia, Adesorji Oderinde, Chinedu Ivonye, Non ∆F 508 Cystic Fibrosis Diagnosed at Age 50 in an African American Male: A Primary Care Perspective, Clinical Practice, Vol. 4 No. 1, 2015, pp. 1-5. doi: 10.5923/j.cp.20150401.01.
Article Outline
- Cystic fibrosis is a multisystem disease characterized by chronic pulmonary infection and bronchiectasis, pancreatic exocrine impairment, and elevated sweat chloride. With advances in and availability of screening tools, increasing number of cases of cystic fibrosis is diagnosed in adulthood. These patients are characterized by high variability in the presentation and severity, in association with identification of a growing number of mild cases. As it is estimated that within the next decade more than half of all individuals with cystic fibrosis (CF) will be aged 18 years or older, primary caregivers are more likely to come across patients with cystic fibrosis [1]. Identification of patients with atypical CF is of particular importance to the primary care physicians because appropriate referral and treatment can substantially affect quality of life and life expectancy [2]. In addition, identification and counseling of family members about this common recessive disorder is important for additional case detection and reproductive planning [2].
1. Case Presentation
- Our patient first presented at age 57 with chronic productive cough, shortness of breath, fever, weight loss and occasional wheezing. He is a non-smoker and has a daughter. His family history is unremarkable except for coronary artery disease in his brother and father, diabetes mellitus with peripheral vascular disease in his father and diabetes mellitus in his mother. His medical history is significant for latent tuberculosis infection which was treated with isoniazid and recurrent admissions for pneumonias. Seven years previously, he was seen by a Pulmonologist at Detroit medical center, Michigan for recurrent productive cough, which was associated with weight loss and shortness of breath. His physical examination was significant for digital clubbing, prolonged expiratory phase and a wide spread rhonchi. A chest X ray [fig 1] revealed a bilateral diffuse reticulonodular opacities with Hyperinflation and central bronchiectasis and his respiratory culture was positive for Staphylococcus aureus and pseudomonas aeruginosa. Patient was appropriately treated and on follow up, his pulmonary function test showed hyperinflation with substantial airway obstruction, normal diffusion capacity, and no change in obstruction following bronchodilator use. A chest computed tomography scan [fig 2] showed pulmonary hyperexpansion with bronchiectatic changes, interstitial thickening and fibrosis. Subsequently a bone Densitometry revealed osteoporosis of the lumbar spine and an Allergic Bronchogenic Pulmonary Aspergillosis (ABPA) Panel was also positive. The patient underwent a sweat chloride testing which was positive and a subsequent genetic analysis showed 711 mutation with deletion in intron 5, both of which were diagnostic of cystic fibrosis.This patient has atypical cystic fibrosis with pancreatic sufficiency and his disease course has been complicated by poor adherence to medication, including respiratory treatments and airway clearance. His sputum is chronically infected with pseudomonas aeruginosa, Alcaligenes xylosoxidans (highly resistant with variable antibiotic susceptibilities), and Methicillin-resistant Staphylococcus aureus and his pulmonary function test [fig 3] has remarkably deteriorated with the recurrent pneumonias over the last 7 years post diagnosis.
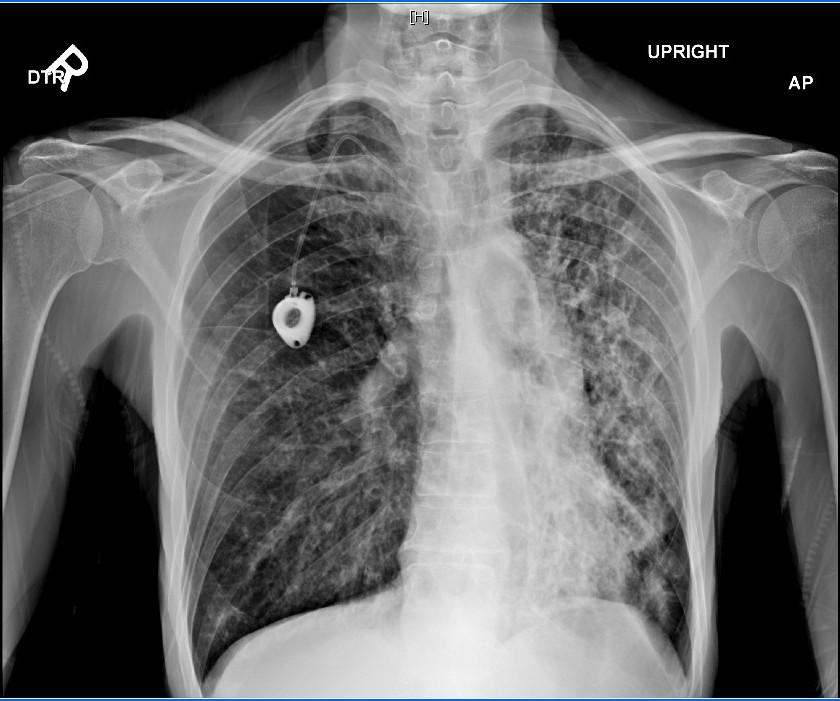 | Figure 1. Chest X ray showing bilateral diffuse reticulonodular opacities and central bronchiectasis |
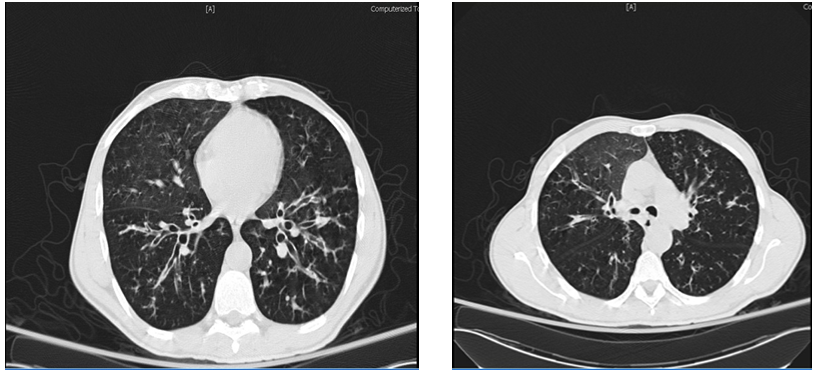 | Figure 2. CT scan showing bronchiectasis, interstitial thickness and fibrosis |
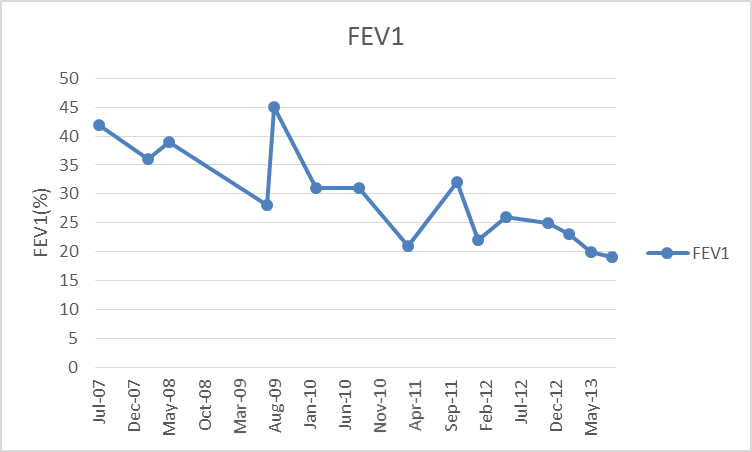 | Figure 3. Forced expiratory volume in the first second (FEV1) from 2007 to 2013 |
2. Discussion
- This patient’s diagnosis of cystic fibrosis was unique. He presented with chronic productive cough, his chest imaging studies showed centralbronchiectasis and hisinitial respiratory culture yielded Pseudomonas aeruginosa and staphylococcus aureus. Subsequent evaluation was significant for a positive ABPA panel and at this point his diagnostic work up could have stopped given the known causal relationship between ABPA and bronchiectasis [3]. However, the presence of osteoporotic changes on his initial imaging studies with bronchiectasis and cystic fibrosis associated pathogen prompted further search. Cystic fibrosis (CF) is a multisystem disease affecting the digestive system, sweat glands, upper and lower respiratory tracts, and the reproductive tract, but progressive lung disease continues to be the major cause of morbidity and mortality [4, 5]. CF is characterized by abnormal transport of chloride and sodium across the respiratory epithelium, resulting in thickened, viscous airway secretions [5, 6]. Over a highly variable time course ranging from months to decades after birth, individuals eventually develop chronic infection of the respiratory tract with a characteristic array of bacterial flora [5, 7], leading to progressive respiratory insufficiency and eventual respiratory failure. The diagnosis of cystic fibrosis requires the presence of cystic fibrosis phenotype and laboratory evidence of Cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction. These include (1) chronic sinopulmonary disease as manifested by chronic cough and sputum production, persistent infection with typical cystic fibrosis pathogens including Staphylococcus aureus, Haemophilusinfluenzae, Pseudomonas aeruginosa, and Burkholderia cepacia, radiographic evidence of bronchiectasis, and chronic sinusitis, often with nasal polyposis; (2) gastrointestinal tract and nutritional abnormalities as manifested by pancreatic insufficiency or recurrent pancreatitis, meconium ileus or distal intestinal obstruction syndrome, failure to thrive or chronic malnutrition, and evidence of focal biliary cirrhosis; and (3) male urogenital problems as manifested by congenital bilateral absence of the vas deferens and obstructive azoospermia [1]. Individuals who demonstrate 1 or more of these features fulfill the criteria for a cystic fibrosis phenotype. But because many of these features are not specific, it is essential that laboratory evidence of CFTR dysfunction is also established before making a diagnosis of cystic fibrosis. Dysfunction in CFTR will have to be demonstrated by a sweat chloride level greater than 60 mEq/L, CFTR genotyping showing the presence of 2 known cystic fibrosis mutations or characteristic bioelectric abnormalities by direct measurement of CFTR function in nasal epithelium [1].
3. Atypical Cystic Fibrosis and the Primary Care Physician
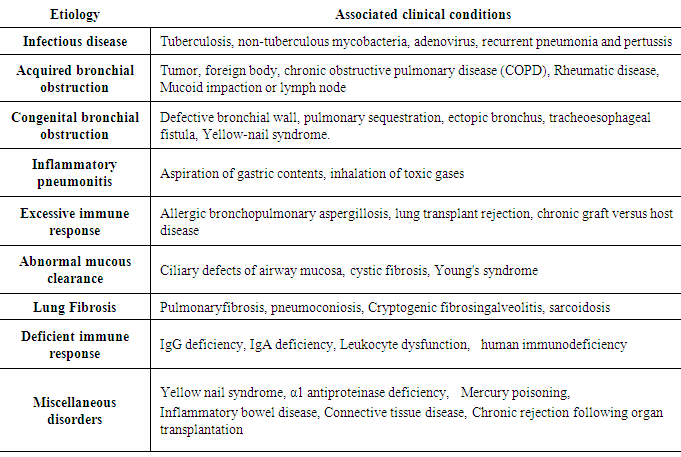
- The increasing number of atypical CF, which represents approximately 2% of affected patients [2], will make clinical diagnosis more challenging. Patients who are first diagnosed with CF as adults are more likely to have atypical features such as normal or intermediate sweat chloride results, one organ system involvement, milder lung disease, and/or little or no gastrointestinal disease [8]. Isolated pulmonary dysfunction with bronchiectasis is quite common among these patients. Table 1, summarized the possible etiological factors associated with bronchiectasis [9].
|
|
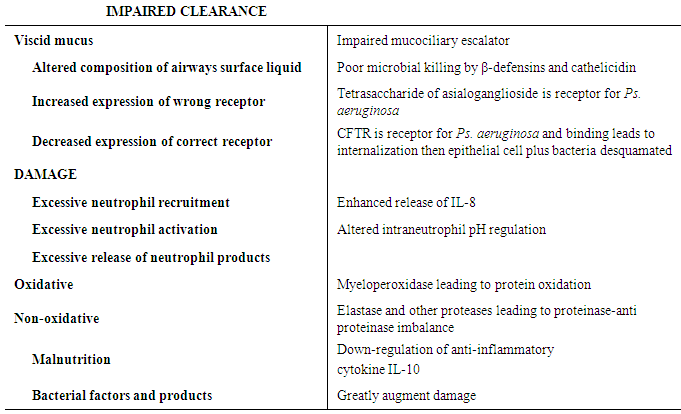
4. Conclusions
- Atypical Cystic fibrosis is an uncommon differential diagnosis in primary care settings and can present as one organ system involvement, mild lung disease, and/or little or no gastrointestinal disease. Prompt identification of atypical cases can substantially improve quality of life and life expectancy in these patients. Screening of family members of individuals who have been diagnosed, including possible prenatal testing should also be offered.