-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Clinical Practice
2012; 1(1): 4-11
doi: 10.5923/j.cp.20120101.02
Maturity-onset Diabetes of the Young (MODY) Genes: Literature Review
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLAttiya K. , Sahar F.
Department of Computer Sciences and Bioinformatics, Mohammad Ali Jinnah University, Islamabad
Correspondence to: Attiya K. , Department of Computer Sciences and Bioinformatics, Mohammad Ali Jinnah University, Islamabad.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
The aim of this article is to give brief introduction about MODY, its different types and to provide a review of the Genetic Screening and Linkage analysis techniques by analyzing the key properties and advance studies presented throughout the particular bibliography, how different MODY genes have been discovered in different populations. The full text articles related to almost all the MODY genes are downloaded. These papers are critically reviewed. The impact of diverse methodology, advantages and limitations of each of the techniques are described. This paper helps us to understand the potency and weak points of different techniques of Linkage analysis and Genetic Screening in argument by highlighting the strengths and weaknesses of these techniques.
Keywords: Keywords MODY- Maturity-Onset Diabetes of the Young, DM-Diabetes Mellitus
Article Outline
1. Introduction
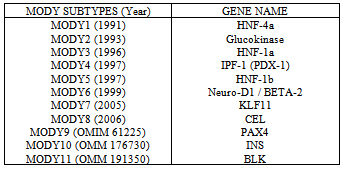
- In humans, diseases are caused either by some microorganisms or due to some mutations in the chromosomes. The diseases which are caused by microorganisms are not inherited (except some viral diseases). But the diseases which are caused by the mutations at gene level are normally heritable and may become lethal. Genetic Disorders arise when one or both copies of a specific gene undergo an alteration known as mutation. Defective genes may also be inherited intact from the parents. Currently about 4,000 genetic disorders are known, with more being discovered. MODY is the term that relies on the elderly classification of diabetes into juvenile-onset and maturity-onset diabetes. An etiology-based classification for diabetes has been revised and introduced by both the American Diabetes Association (ADA) and the World Health Organization (WHO). The group of “Genetic defect in B-cell function” has now included MODY as its part with its sub classification according to the gene involved Martine et al. (2008). Tattersall (U.K.) and Fajan (U.S.A.) first described MODY in 1974 Tattersal (1975), after taking into account a group of young people with diabetes who were treated without insulin 2 years or more after diagnosis. Since the 1970s there has been great interest in MODY as it is a genetic form of diabetes. MODY is an ancestral form of early-onset type2 diabetes. It is a monogenic form of diabetes mellitusinherited in autosomal dominant mode (Anna, et al. (2001) and Owen, et al. (2001)). It is primarily an outcome of impaired B-Cells of pancreas (Anna, et al. (2001) and Owen, et al. (2001)). MODY is not a single entity but represents genetic, metabolic, and clinical heterogeneity Costa1, et al. (2000). MODY generally develops in middle age, and mainly coupled with primarily scantiness of insulin secretion Vaxillaire, et al. (2006). As MODY is a monogenic form of diabetes, most monogenic diabetes genes are fundamentally B-cell genes. A key outcome has been that the immense mass of genes where mutations cause early-onset diabetes have condensed B-cell function rather than improved insulin confrontation. Till 2009 eight discrete MODY genes have been acknowledged Maciej, et al. (2009). These are the genes including HNF4A, encoding hepatocyte nuclear factor 4 Alpha Yamagata, et al. (1996), GCK, encoding glucokinase Froguel, et al. (1993), HNF1A, encoding hepatocyte nuclear factor 1 Alpha Yamagata, et al. (1996), IPF1, encoding insulin promoter factor 1 Stoffers, et al. (1997), HN F1B, encoding hepatocyte nuclear factor 1 Beta Horikawa, et al. (1997), NEUROD1, encoding neurogenic differentiation 1 Malecki, et al. (1999) and KLF11, encoding for kruppel-like factor 11 Neve, et al. (2005) and now four further genes have been exposed that cause MODY. These genes are PAX4, encoding, Paired Domain gene 4 (OMIM 612225), CEL, encoding carboxyl-ester lipase Raeder, et al. (2006), INS, encoding, Insulin (OMIM 176730) and BLK, encoding, Tyrosine kinase B-Lymphocyte. (OMIM 191305)
2. Overview
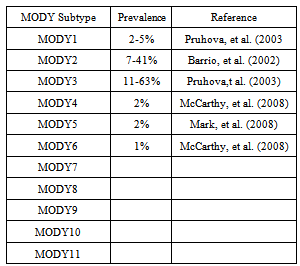
- Molecular genetic studies of MODY families have demonstrated that this clause is not a solo unit but is a clinically and hereditarily heterogeneous syndrome Martine, et al. (2008). Mutations, deletions, or other anomalies in at least eleven genes are a root for the bulk of the MODY cases R.Pearson, et al. (2006). These MODY genes encode the enzymes glucokinase (GCK (MODY 2)) that is liable for the early processing of glucose in the B-cell (Doria, et al. (2000) and Sung, et al. (2004)) and copious transcription factors that modulate the expression of numerous genes concerned with the demarcation and utility of B-cells (Doria, et al. (2000) and Sung, et al. (2004)).The first MODY gene to be documented was glucokinase (GCK) Froguel, et al. (1992) and Hattersley, et al. (1992), followed by hepatocyte nuclear factors HNF1A (TCF1), HNF4A Yamagata, et al. (1996) and others.The table 1 shows the genetic cataloging and detached phenotypes of the MODY subtypes.
3. Different Subtypes of Mody
- HNF4A SUBTYPE (OMIM 125850)MODY type 1 is caused by mutation in Hepatocyte Nuclear Factor 4 alpha (HNF4A) gene. This gene is also known as NR2A1 (nuclear receptor subfamily 2, group A, member 1). It is a nuclear recopter encoded by HNF4A gene (Chartier, et al. (1994) and Argyrokastritis, et al. (1997)). This gene is located on chromosome 20. HNF4A is responsible for the regulation of hepatic and pancreatic ß cell gene expression (Sladek, et al. (1990) and Kuo, et al. (1992)). Heterozygous mutation in human HNF4A gene results in progressive decrease in insulin production Yamagata, et al. (1996).Heterozygous mutations in HNF4A are considered a rare source of MODY compared with MODY2/ GCK and MODY3/HNF1A mutations (Matschinsky (2005) and Pearson, et al. (2005). HNF4-A belongs to the steroid/thyroid hormone receptor super family. This gene plays a role in development of the liver, kidney, and intestines. HNF4A mutations are associated with an increase in birth weight and macrosomia and thus can be viewed as a novel cause of neonatal hypoglycemia Pearson, et al. (2007). GCK SUBTYPE (OMIM 125851)Glucokinase (GCK) is also called hexokinase IV or D. Heterozygous mutations of the glucokinase gene (GCK) result in MODY type 2 Froguel, et al. (1994). This gene maps to chromosome 7. It is the most common form of MODY Matschinsky, et al. (1990). Unlike other MODY subtypes, the pathophysiology involves impaired glucose sensing by the pancreatic β cells, resulting in mild non-progressive hyperglycemia (5.5 – 8 m mol/L) that is often asymptomatic Ohn, et al. (2009).
|

|
4. Literature Review
- Genetic studies are contributing much in establishing the linkage association for genetic disorders and have been employed widely in identification of diseased genes. Different types of MODY have been discovered in European populations but still MODY is misdiagnosed as T2DM. MODY genes are named as MODY 1 to MODY 11 according to the year they have recognized in MODY patients. The table below shows the eleven subtypes of MODY, their gene name and the year they were recognized.The primary objective of this chapter is to review the literature on the methodologies of the planned research area. This chapter will aid to gain background knowledge of the proposed research topic. It will confer the work that has been done up till now related to genetic screening and linkage analysis by different scientists/biologists. A thorough survey on preprocessing methodologies will facilitate us to not only compare and contrast the previous work but also with the proposed work. We will find out the problems in the area of interest that have already been investigated. Further, we will identify the potential sources of information for conducting the detailed research on proposed work.
|
4.1. Review of Linkage Analysis/Genetic Screening Methodologies
- Although there has been significant amount of research in the area of Diabetes and particularly on Maturity-onset diabetes of the young. But the work that is most related to the current research study with respect to methodologies is as follows:Fajans (1989) performed the linkage analysis of a family that consists of 360 members spanning 6 generations and 74 members with diabetes, including those with MODY. This family had been studied prospectively since 1958. Linkage studies showed that the gene responsible for MODY in this family is tightly linked to 20q12-q13.1. This gene that maps to this chromosome is known as HNF1A. However, Fajans did not screen the HNF1A for detection of new mutation. Similarly Froguel, (1992) also did linkage analysis on 16 French families with maturity-onset diabetes of the young and found that the linkage of the disease with GCK. There was statistically significant evidence of genetic heterogeneity, with an estimated 45 to 95% of the 16 families showing linkage to glucokinase. Because glucokinase is a key enzyme of blood glucose homeostasis, the results suggested a pathogenetic connection. The demonstration by Fajans (1989), that the gene for HNF1A is the site of mutations causing MODY3 prompted Yamagata, et al. (1996) to screen the gene for HNF4A for a mutation.Yamagata, et al. (1996) verified a gln268-to-ter mutation in the gene encoding hepatocyte nuclear factor-4-alpha in a multigenerational family, in which type I maturity-onset diabetes of the young was first defined. HNF-4-alpha is most highly expressed in liver, kidney, and intestine. It is also expressed in pancreatic islets and insulinoma cells. It is a key regulator of hepatic gene expression and is a major activator of HNF-1-alpha (TCF1), which in turn activates the expression of a large number of liver-specific genes, including those involved in glucose, cholesterol, and fatty acid metabolism. This shows that these genes are somehow linked to each other. This association of genes will be determined through association rule mining techniques. Vixillaire, et al., (1997) examined 10 unrelated Caucasian families in whom MODY cosegregated with markers for MODY3 and found 10 different mutations in the TCF1 gene, all of which cosegregated with diabetes. In these families, they found no obvious relationships between the nature of the mutations observed (i.e., frame shift, nonsense, or missense), or their location in the gene, with clinical features of diabetes (e.g., age at onset, severity). Vixillaire research is limited to only affected members of MODY. He did not study the normal member to differentiate the wild and mutant genes in these families. Costa, et al. (2000) investigate the frequencies of the major MODY subtypes in a panel of Spanish families. They also assessed the phenotypic difference in patients with different MODY subtypes. He concluded that mutations in MODY2 (GCK) and MODY 3 (HNF1A) genes contribute much in causing MODY in majority of the cases of Spanish MODY families. They wind up the results with the knowledge that the relative frequencies of MODY genes are in agreement with already reported prevalence of these genes in European populations.Hansen, et al. (2000) examined 200 Danish patients with late-onset type II diabetes and 44 Danish and Italian MODY patients for mutations in the IPF1 gene by SSCP and heteroduplex analysis. In the patients with late-onset type II diabetes, they identified a noncoding G insertion/deletion polymorphism at nucleotide -108, a silent gly54-to-gly substitution, and the rare D76N variant. Moreover, a Danish MODY patient carried an ala140-to-thr (A140T) variant. Neither the D76N nor the A140T variant segregated with diabetes, and their transcriptional activation of the human insulin promoter expressed in vitro was indistinguishable from that of wild type. Johansen, et al. (2005) examined the prevalence and nature of mutations in the 3 common MODY genes HNF4A, GCK, and TCF1 (HNF1A) through genetic screening in Danish patients with a clinical diagnosis of MODY and determined metabolic differences in probands with and without mutations in HNF4A, GCK, and TCF1. They identified 29 different mutations in 38 MODY families. Their findings suggested a relative prevalence of 3% of MODY1 (2 different mutations in 2 families), 10% of MODY2 (7 in 8), and 36% of MODY3 (21 in 28) among Danish kindred clinically diagnosed as MODY. Pinterove, et al. (2007) screened the GCK gene that was known to cause MODY 2 in Froguel (1992). He examined the 92 Czech probands fulfilling classic MODY criteria and identified 15 different missense mutations in 27 (29%) patients; the mutations were not found in 50 unrelated healthy Czech individuals. Pinterove, et al. (2007) concluded that mutations in GCK are a common cause of MODY in the Czech population. Pakistan is a developing country and not free from health burden threatened by MODY. It is even more difficult to diagnose and classify MODY in the country as no genetic research has been done so far. The prospective basis of above mentioned work done on linkage analysis and genetic screening of MODY will help us to conduct detailed research on linkage analysis and genetic screening of MODY in Pakistani population. The summary of the methodologies adopted by different researchers that did examination on MODY patients and their experimental results are briefly shown in the table 4:
6. Discussion
- In this chapter we have discussed the work that has been done so far in the context of genetic screening and association extraction. Till now eleven genes contributing to MODY have been discovered in European population. These genes have been screened and linked in different populations by different scientists and researchers to find out new diseased gene location on chromosomal loci.
7. Summary
- This section describes the key points of this review article.i. This article is a literature review of Genetic screening and linkage analysis of MODY genes, an application of Bioinformatics, for the discovery of new diseased genes and mutations.ii. This article will help the researchers and scientists to carry out research in this context to detect new genes in different populations.
ACKNOWLEDGMENTS
- I would like to say thanks to my supervisor Dr. Sahar Fazal for her indispensable moral support in this article.
Disclosure
- Both the authors contributed equally in this review article.
References
| [1] | Anna L.Gloyn, Sian Ellard, Maggie Shepherd, Rodney T. Howell, Elizabeth M.Parry, Andrew Jefferson, Elaine R. Levy And Andrew T. Hattersley: Maturity-Onset Diabetes Of The Young Caused By A Balanced Translocation Where The 20q12 Break Point Results In Disruption Upstream Of The Coding Region Of Hepatocyte Nuclear Factor-4A Gene. |
| [2] | Tattersall RB, Fajans SS: A Difference Between The Inheritance Of Classical Juvenile-Onset And Maturity-Onset Type Diabetes Of Young People. Diabetes, 1975 Jan;24(1):44-53 |
| [3] | Owen K, Httersley A: Maturity-Onset Diabetes Of The Young: From Clinical Description To Molecular Genetic Characterization. In Best Practice And Research Clinical Endocrinology And Metabolism. Vol. 15. Dunger D, Bains S, Eds., London, Harcourt, 2001, P. 309-323 |
| [4] | A Costa1, M Besco´S2, G Velho3, J C CheˆVre4, J Vidal1, G Sesmilo1, C Bellanne´-Chantelot5, P Froguel4, R Casamitjana2, F Rivera-Fillat2, R Gomis1 And I Conget1: Genetic And Clinical Characterisation Of Maturity-Onset Diabetes Of The Young In Spanish Families. European Journal Of Endocrinology (2000) 142 380–386 |
| [5] | Vaxillaire M, Froguel P 2006 Genetic Basis Of Maturity-Onset Diabetes Of The Young. Endocrinol Metab Clin North Am 35:371–384 |
| [6] | D. Michie, D. J. Spiegelhalter, And C. C. Taylor: Machine Learning, Neural And Statistical Classification. London, U.K.: Ellis Horwood, 1994. |
| [7] | Yamagata, K., Furuta, H., Oda, N., Kalsaki, P. J., Menzel, S., Cox, N. J., Fajans, S. S., Signorini, S., Stoffel, M., Bell, G. I. (1996). Mutations In The Hepatocyte Nuclear Factor-4-Alpha Gene In Maturity-Onset Diabetes Of The Young (MODY1). Nature 384: 458-460. |
| [8] | Froguel P, Et Al. (1993) Familial Hyperglycemia Due To Mutations In Glucokinase. Definition Of A Subtype Of Diabetes Mellitus. N Engl J Med 328:697–702. |
| [9] | Yamagata K, Et Al. (1996) Mutations In The Hepatocyte Nuclear Factor-1alpha Gene In Maturity-Onset Diabetes Of The Young (MODY3). Nature 384:455–458. |
| [10] | Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic Agenesis Attributable To A Single Nucleotide Deletion In The Human IPF1 Gene Coding Sequence. Nat Genet. 1997;15:106–10. |
| [11] | Horikawa Y, Et Al. (1997) Mutation In Hepatocyte Nuclear Factor-1 Beta Gene (TCF2) Associated With MODY. Nat Genet 17:384–385. |
| [12] | Malecki MT, Et Al. (1999) Mutations In NEUROD1 Are Associated With The Development Of Type 2 Diabetes Mellitus. Nat Genet 23:323–328. |
| [13] | Neve B, Et Al. (2005) Role Of Transcription Factor KLF11 And Its Diabetes-Associated Gene Variants In Pancreatic Beta Cell Function. Proc Natl Acad Sci USA 102:4807–4812. |
| [14] | Martine Vaxillaire And Philippe Froguel: Monogenic Diabetes In The Young, Pharmacogenetics And Relevance To Multifactorial Forms Of Type 2 Diabetes. Endocrine Reviews, May 2008, 29(3):254–264 |
| [15] | Pearson ER, Pruhova S, Tack CJ Johansen A, Castleden HA, Lumb PJ, Wierzbicki AS, Clark PM, Lebl J, Pedersen O, Ellard S, Hansen T, Hattersley AT 2005 Molecular Genetics And Phenotypic Characteristics Of MODY Caused By Hepatocyte Nuclear Factor 4_ Mutations In A Large European Collection. Diabetologia 48:878–885 |
| [16] | Sung-Hoon Kim,1,3 Xiaowei Ma,1,3 Stanislawa Weremowicz,2,4 Tonino Ercolino,1,3 Christine Powers,1 Wojciech Mlynarski,1,3 K. Aviva Bashan,1 James H. Warram,1 Josyf Mychaleckyj,5 Stephen S. Rich,5 Andrzej S. Krolewski,1,3 And Alessandro Doria1,3 Identification Of A Locus For Maturity-Onset Diabetes Of The Young On Chromosome 8p23 DIABETES, VOL. 53, MAY 2004 |
| [17] | Doria A, Plengvidhya N: Recent Advances In The Genetics Of Maturity Onset Diabetes Of The Young And Other Forms Of Autosomal Dominant Diabetes. Curr Opin Endocrinol Diabetes 7:203–210, 2000 |
| [18] | Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO, Lesage S, Vionnet N, Clement K, Fougerousse F, Et Al.: Close Linkage Of Glucokinase Locus On Chromosome 7p To Early-Onset Non-Insulin-Dependent Diabetes Mellitus. Nature 356:162–164, 1992 |
| [19] | Hattersley AT, Turner RC, Permutt MA, Patel P, Tanizawa Y, Chiu KC, O’Rahilly S, Watkins PJ, Wainscoat JS: Linkage Of Type 2 Diabetes To The Glucokinase Gene. Lancet 339:1307–1310, 1992 |
| [20] | Argyrokastritis A, Kamakari S, Kapsetaki M, Kritis A, Talianidis I, Moschonas NK (February 1997). "Human Hepatocyte Nuclear Factor-4 (Hhnf-4) Gene Maps To 20q12-Q13.1 Between PLCG1 And D20S17". Hum.Genet.99 (2):23 6.Doi:10.1007/S004390050345.PMID 9048927. |
| [21] | Chartier FL, Bossu JP, Laudet V, Fruchart JC, Laine B (September 1994). "Cloning And Sequencing Of Cdnas Encoding The Human Hepatocyte Nuclear Factor 4 Indicate The Presence Of Two Isoforms In Human Liver".Gene 147 (2):269–72. Doi:10.1016/0378-1119(94)90079-5. PMID 7926813. |
| [22] | Kuo CJ, Conley PB, Chen L, Sladek FM, Darnell JE, Jr, Crabtree GR. A Transcriptional Hierarchy Involved In Mammalian Cell-Type Specification. Nature. 1992;355:457–61. |
| [23] | Sladek FM, Zhong WM, Lai E, Darnell JE., Jr Liverenriched Transcription Factor HNF-4 Is A Novel Member Of The Steroid Hormone Receptor Superfamily. Genes Dev. 1990;4:2353–65. |
| [24] | Matschinsky FM 2005 Glucokinase, Glucose Homeostasis, And Diabetes Mellitus. Curr Diab Rep 5:171–176 |
| [25] | Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, Ellard S, Ferrer J, Hattersley AT 2007 Macrosomia And Hyperinsulinaemic Hypoglycaemia In Patients With Heterozygous Mutations In The HNF4A Gene. Plos Med 4:E118 |
| [26] | Froguel P, Velho G. Maturity-Onset Diabetes Of The Young. Curr Opin Pediatr. 1994;6:482–5. |
| [27] | Matschinsky FM. Glucokinase As Glucose Sensor And Metabolic Signal Generator In Pancreatic Beta-Cells And Hepatocytes. Diabetes. 1990;39:647–52. |
| [28] | Ohn Nyunt,*1 Joyce Y Wu,2 Ivan N Mcgown,2 Mark Harris,1 Tony Huynh,1 Gary M Leong,1 David M Cowley,2 And Andrew M Cotterill1 Investigating Maturity Onset Diabetes Of The Young Clin Biochem Rev. 2009 May; 30(2): 67–74. |
| [29] | Fajans SS, Bell GI, Polonsky KS 2001 Molecular Mechanisms And Clinical Pathophysiology OfMaturity-Onset Diabetes Of The Young. N Engl J Med 345:971–980 |
| [30] | Vaxillaire M, Froguel P 2006 Genetic Basis Of Maturity-Onset Diabetes Of The Young. Endocrinol Metab Clin North Am 35:371–384 |
| [31] | Velho G, Hattersley AT, Froguel P 2000 Maternal Diabetes Alters Birth Weight In Glucokinase-Deficient (MODY2) Kindred But Has No Influence On Adult Weight, Height, Insulin Secretion Or Insulin Sensitivity. Diabetologia 43:1060–1063 |
| [32] | Hattersley AT, Beards F, Ballantyne E, Appleton M, Harvey R, Ellard S 1998 Mutations In The Glucokinase Gene Of The Fetus Result In Reduced Birth Weight. Nat Genet 19:268–270 |
| [33] | Stride A, Ellard S, Clark P Shakespeare L, Salzmann M, Shepherd M, Hattersley AT 2005 _-Cell Dysfunction, Insulin Sensitivity, And Glycosuria Precede Diabetes In Hepatocyte Nuclear Factor-1_ Mutation Carriers. Diabetes Care 28:1751–1756 |
| [34] | Maestro MA, Cardalda C, Boj SF, Luco RF, Servitja JM, Ferrer J 2007 Distinct Roles Of HNF1 _, HNF1 _, And HNF4 _ In Regulating Pancreas Development, _-Cell Function And Growth. Endocr Dev 12:33–45 |
| [35] | Haumaitre C, Barbacci E, Jenny M, Ott MO, Gradwohl G, Cereghini S 2005 Lack Of TCF2/Vhnf1 In Mice Leads To Pancreas Agenesis. Proc Natl Acad Sci USA 102:1490–1495 |
| [36] | Lindner TH, Cockburn BN, Bell GI 1999 Molecular Genetics Of MODY In Germany. Diabetologia 42:121–123 |
| [37] | Poulin G, Turgeon B, Drouin J (November 1997). "Neurod1/Beta2 Contributes To Cell-SpecificTranscription Of The Proopiomelanocortin Gene". Mol. Cell. Biol.17 (11): 6673–82. PMID 9343431.PMC 232521. |
| [38] | Bellanne-Chantelot C, Chauveau D, Gautier JFDubois-Laforgue D, Clauin S, Beaufils S, Wilhelm JM, Boitard C, Noe¨L LH, Velho G, Timsit J 2004 Clinical Spectrum Associated With Hepatocyte Nuclear Factor-1_ Mutations. Ann Intern Med 140:510–517 |
| [39] | Naya FJ, Huang HP, Qiu Y, Mutoh H, Demayo FJ, Leiter AB, Et Al. Diabetes, Defective Pancreatic Morphogenesis, And Abnormal Enteroendocrine Differentiation InBETA2/Neurod-Deficient Mice. Genes Dev. 1997;11:2323–34. |
| [40] | Neve B, Et Al. (2005) Role Of Transcription Factor KLF11 And Its Diabetes-Associated Gene Variants In Pancreatic Beta Cell Function. Proc Natl Acad Sci USA102:4807–4812. |
| [41] | Scohy S, Gabant P, Van Reeth T, Hertveldt V, Dreze PL, Van Vooren P, Riviere M, Szpirer J, Szpirer C (Jan 2001). "Identification Of KLF13 And KLF14 (SP6), Novel Members Of The SP/XKLF Transcription Factor Family". Genomics 70 (1):93–101. Doi:10.1006/Geno.2000.6362.PMID 11087666. |
| [42] | Cook T, Gebelein B, Mesa K, Mladek A, Urrutia R (Nov 1998). "Molecular Cloning And Characterization Of TIEG2 Reveals A New Subfamily Of Transforming Growth Factor-Beta-Inducible Sp1-Like Zinc Finger-Encoding Genes Involved In The Regulation Of Cell Growth". J Biol Chem 273 (40): 25929–36.Doi:10.1074/Jbc.273.40.25929.PMID 9748269. |
| [43] | Matsushita T, Yamaoka T, Otsuka S, Moritani M, Matsumoto T, Itakura M (January 1998). "Molecular Cloning Of Mouse Paired-Box-Containing Gene (Pax)-4 From An Islet Beta Cell Line And Deduced Sequence Of Human Pax-4". Biochem. Biophys. Res. Commun. 242 (1): 176–80. Doi:10.1006/Bbrc.1997.7935. PMID 9439631. |
| [44] | Inoue H, Nomiyama J, Nakai K, Matsutani A, Tanizawa Y, Oka Y (February 1998). "Isolation Of Full-Length Cdna Of Mouse PAX4 Gene And Identification Of Its Human Homologue". Biochem. Biophys. Res. Commun. 243 (2): 628–33. Doi:10.1006/Bbrc.1998.8144. PMID 9480859. |
| [45] | Bell GI, Pictet RL, Rutter WJ, Cordell B, Tischer E, Goodman HM (March 1980). "Sequence Of The Human Insulin Gene". Nature 284 (5751):26–32. Doi:10.1038/284026a0. PMID 6243748. |
| [46] | Dandona, P., Aljada, A., Mohanty, P., Ghanim, H., Hamouda, W., Assian, E., Ahmad, S. Insulin Inhibits Intranuclear Nuclear Factor Kappa-B And Stimulates I-Kappa-B In Mononuclear Cells In Obese Subjects: Evidence For An Anti-Inflammatory Effect? J. Clin. Endocr. Metab. 86: 3257-3265, 2001.[Pubmed: 11443198] |
| [47] | Drebin JA, Hartzell SW, Griffin C, Campbell MJ, Niederhuber JE (Mar 1995). "Molecular Cloning And Chromosomal Localization Of The Human Homologue Of A B-Lymphocyte Specific Protein Tyrosine Kinase(Blk)".Oncogene 10 (3): 477–86. PMID 7845672. |
| [48] | Dymecki, S., Niederhuber, J., Desiderio, S. Specific Expression Of A Novel Tyrosine Kinase Gene, Blk, In B Lymphoid Cells. Science 247: 332-336, 1990.[Pubmed: 2404338] |
| [49] | Barrio R, Bellanne-Chantelot C, Moreno JC, Morel V, Calle H, Alonso M, Mustieles C 2002 Nine Novel Mutations In Maturity-Onset Diabetes Of The Young (MODY) Candidate Genes In 22 Spanish Families. J Clin Endocrinol Metab 87:2532–2539 |
| [50] | Mark I, Mccarthy And Andrew T. Hattersley: Novel Insights Arising From The Definition Of Genes Fro Monogenic And Type 2 Diabetes. DIABETES, VOL. 57, NOVEMBER 2008 |
| [51] | Pruhova S, Ek J, Lebl J, Sumnik Z, Saudek F, Andel M, Pedersen O, Hansen T 2003 Genetic Epidemiology Of MODY In The Czech Republic: New Mutations In The MODY Genes HNF-4_, GCK And HNF-1_. Diabetologia 46:291–295 |
| [52] | Fajans, S. S. (1989). Maturity-Onset Diabetes Of The Young (MODY). Diabetes Metab. 5: 579-606. |
| [53] | Froguel, P., Vaxillaire, M., Sun, F., Velho, G., Zouali, H., Butel, M. O., Lesage, S., Vionnet, N., Clement, K., Fougerousse, F., Tanizawa, Y., Weissenbach, J., Beckmann, J. S., Lathrop, G. M., Passa, P., Permutt, M. A., Cohen, D. (1992). Close Linkage Of Glucokinase Locus On Chromosome 7p To Early-Onset Non-Insulin-Dependent Diabetes Mellitus. Nature 356: 162-164. |
| [54] | Vaxillaire, M., Rouard, M., Yamagata, K., Oda, N., Kaisaki, P. J., Boriraj, V. V., Chevre, J.-C., Boccio, V., Cox, R. D., Lathrop, G. M., Dussoix, P., Philippe, J., Timsit, J., Charpentier, G., Velho, G., Bell, G. I., Froguel, P. (1997). Identification Of Nine Novel Mutations In The Hepatocyte Nuclear Factor 1 Alpha Gene Associated With Maturity-Onset Diabetes Of The Young (MODY3). Hum. Molec. Genet. 6: 583-586. |