-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Computer Science and Engineering
p-ISSN: 2163-1484 e-ISSN: 2163-1492
2017; 7(1): 22-27
doi:10.5923/j.computer.20170701.03

Comparative Analysis of Chromosomal Variation Altered New Region for Detecting Biomarker in Liver Cancer
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLEsraa M. Hashem1, Mai S. Mabrouk1, Ayman M. Eldeib2
1Biomedical Engineering Department, Misr University for Science and Technology (MUST University), Egypt
2Systems and Biomedical Engineering, Faculty of engineering, Cairo University, Giza, Egypt
Correspondence to: Mai S. Mabrouk, Biomedical Engineering Department, Misr University for Science and Technology (MUST University), Egypt.
| Email: |  |
Copyright © 2017 Scientific & Academic Publishing. All Rights Reserved.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Liver cancer is one of the most widely recognized malignancies in adults, hepatocellular carcinoma (HCC) is an out face malignancy. Nearly instances of HCC are identified with a preceding stage, and, in this manner, medication choices are restricted. Most Hepatic carcinoma emerge from the accumulation of genetic abnormalities and potentially induced by exterior etiological factors. Genome-wide gene expression profiling is a generally utilized system for discovering new potential biomarkers for prediction and diagnosis of disease severity. In this work, two statistical techniques (circular binary segmentation and discrete stationary wavelet transform) are utilized for detection of copy number alterations to test single-nucleotide polymorphism array of ten cell lines hepatic tumor patients on certain chromosomes, which are frequently detected in hepatic cancer have been proposed. Results detect a new mutated chromosomal amplification region (4q22.1) has been identified through our investigation utilizing both methods. This region sets up, SPP1, and AFF1 tumor suppressor genes and has not observed previously, that opening new frontiers that can be involved in liver carcinogenesis.
Keywords: Hepatocellular carcinoma, Circular binary segmentation, Discrete stationary wavelet transform, Single-nucleotide polymorphism
Cite this paper: Esraa M. Hashem, Mai S. Mabrouk, Ayman M. Eldeib, Comparative Analysis of Chromosomal Variation Altered New Region for Detecting Biomarker in Liver Cancer, Computer Science and Engineering, Vol. 7 No. 1, 2017, pp. 22-27. doi: 10.5923/j.computer.20170701.03.
Article Outline
1. Introduction
- Cancer has been over and again distinguished as a controlled disease. Certainly, the procedure of tumor growth from primary to malignant structures, including metastasis to different cells includes the conflict between various cell lineages contend to adjust to the changing conditions both inside and around the tumor. This complex operation is almost associated with genetic alterations and the incidence of mutations [1]. Greater than 80% of hepatic cancers are hepatocellular carcinoma (HCCs), It is once in a while identified early [2]. The end stage of many different chronic liver diseases is liver cirrhosis, it is normally diagnosed at an end-stage when effective treatment is impossible. Patient with cirrhosis have an over risk of liver tumor. Mostly cirrhosis created because of: hepatitis C (HCV), alcoholic liver disease (ALD), hepatitis B (HBV), auto-immune hepatitis (AIH), and other risk factors [3], as shown in Figure 1-1. As stated by those current studies, the large part of liver cancer patients contracted the disease from the accumulation the abnormalities of genetics, possibly rise by exterior etiological factors especially HCV and HBV contaminations [4]. So far, no significant proof has found that enhances survival utility with surveillance of high-risk patients. The diagnostic tools which utilized commonly to attempt to enhance patient survival are: biomarkers, liver biopsy, radiographic imaging and serum tumor marker alpha-fetoprotein (AFP) [5].
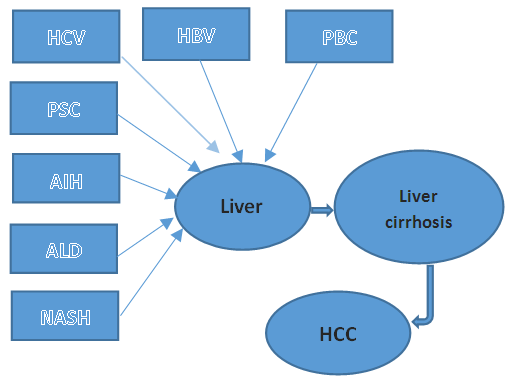 | Figure 1.1. Causes of liver cirrhosis |
2. Materials and Methods
- The most profuse genetic variants are SNPs, which supply the ultimate comprehensive resource for verification of genetic diversity [17]. The progress in high-throughput sequencing technology has made copy number discovery much easier, the application of known CNA information means that we can target structural variation in a sample using less expensive techniques such as the SNP array without a large reduction in genome-wide coverage.
2.1. Experimental Setup
- The data used in this study is SNP Genotyping Array, performed on Human Duov1 DNA investigation Bead Chip (HumanCNV370v1), includes the usual SNP content items on a HumanHap300-Duo Genotyping Bead Chip, with an extra 52,167 marker intended to specifically target close-by 14,000 CNA areas of the genome, for an aggregate of more than 370,000 markers. Ten HCC cell lines process according to the manufacturers' instructions. Raw data processed utilizing an in-house pipeline to take out a segment of copy number and gene-summarized copy number assessments. The data on the normalized log R ratios are obtainable in GEO [http://www.ncbi.nlm.nih.gov/geo] with accession number [GSE38207].
2.2. Statistical Methods
- Circular Binary Segmentation (CBS):Because of the complication of genetic variations in tumor cells, high density SNP array investigation is an extremely requesting method in the field of cancer research. The implementation of SNP arrays in combination with tissue array, PCR and functional analysis advances will finally prompt to the full revelation of the complex genetic alterations in human cancers.CBS can give a characteristic approach to segment a chromosome into contiguous regions iteratively searching for parts of chromosomal regions and bypasses parametric modeling of the data with its utilize of a variation reference division. Once the chromosome partitioned, a copy number can appraise from the segments and discover the change points on SNP array data with the help of assistance information such as the ploidy of the chromosome [18]. The algorithm stops when no more statistically significant change-points are situated, according to a heuristically chosen significance threshold. This information will give the locations of copy number aberrations, assigned by Zi as appeared in (1). Where
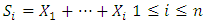 are the partial sums and
are the partial sums and  is the log R ratios of the intensities. The probability proportion measurement for checking the null hypothesis that there is no change against the alternative that there is exactly one change at an unknown location i is given by (2) [19].
is the log R ratios of the intensities. The probability proportion measurement for checking the null hypothesis that there is no change against the alternative that there is exactly one change at an unknown location i is given by (2) [19].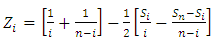 | (1) |
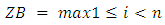 | (2) |
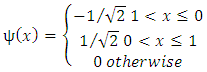 | (3) |
3. Results
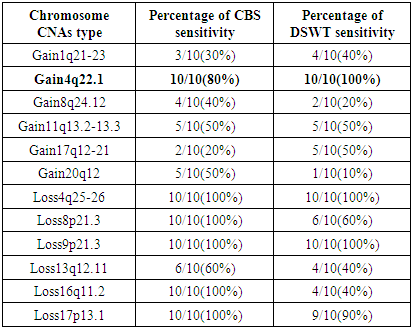
- HCC is a standout amongst the ultimate recurrent cancer entities worldwide, Hepatitis C and Hepatitis B are the main underlying basic causes of chronic liver disease which leads to cirrhosis. The tumor recurrence is the major restriction of long survival after liver transplantation. Most recent advancement of technologies for gene expression profiling and genome copy number investigation enable the comprehensive characterization of entire cancer genomes. This information will enhance our comprehension of the epigenetic and genetic aberrations leading to the evolution of HCC, as well as ultimately to provide information that helps in the treatment of patients [24].By performing CBS algorithm in HCC cell lines. A region of 4q25-26 losses observed in 10 out of 10-HCC (100%), the allelic loss of chromosome 4q were significantly associated with HCC has elevated serum AFP [25]. A loss of 8p21.3 was observed in 10 out of 10-HCC (100%), loss of 9p was observed in 10 out of 10-HCC (100%) also loss of 16q11.2 and 17p13.1 observed in 10 out of 10-HCC (100%), all the results are summarized in table 3.1. While performing DSWT algorithm, it has been noticed that, the most commonly gained region of chromosome 1q22.1–23.1 in 4 out of 10-hepatocellular carcinoma (40%). Gains of 1q are the most frequent variation and occur early during tumor-genesis [26]. This region contains the tumor suppressors genes JTB, CCT3and COPA [27]. A loss of 4q was detected in 10 out of 10-HCC (100%). Loss for 9p additionally observed in 10 out of 10-HCC (100), the results summarized in table 3.1. This finding agrees with those of previous studies [28] leading to the assumption that these aberrations could have an essential part in the preparation of HCC, probably based on variations of genes lying on these chromosome segments.
|
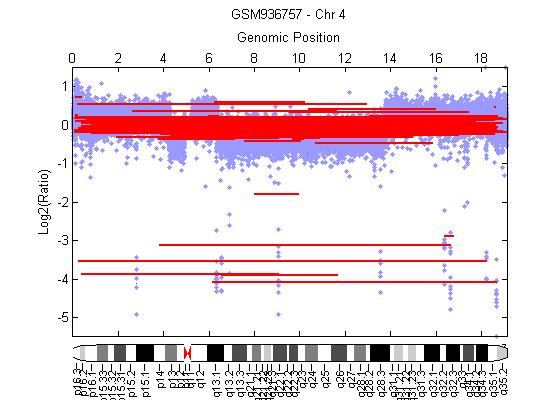 | Figure 3.1. Gain of chromosome 4q22.1 using CBS algorithm |
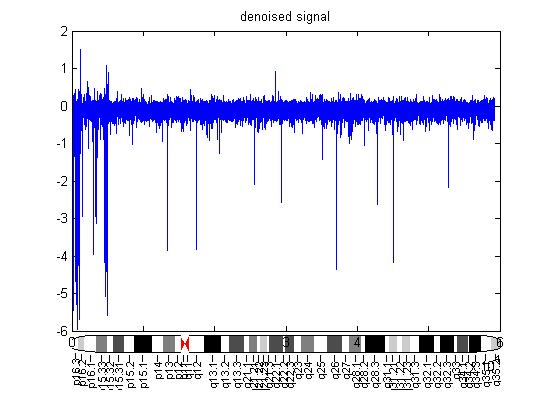 | Figure 3.2. Gain of chromosome 4q22.1 using DSWT |
4. Discussions
- The liver has many identified functions related to metabolism, processing, and immunity, the liver disease indicates a different number of concerns for the delivery of medical consideration. Large-scale CN changes in the genomes of humans have been disregarded mostly because of technical challenges in scoring these changes genomes-wide with a favorable resolution.Gene expression profiling can prompt to discover new markers that may help enhance diagnostic for detect HCC, represent novel targets for therapeutic agents, and permit us to identify regions in the HCC genome that have undergone recurrent high-level focal amplifications or deletions. It should be considered for future investigation on detecting CNA regions.
5. Conclusions
- Hepatic carcinoma is a tumor of the liver, which is generally emerging in the background of chronic liver diseases. The assessment for patients with liver cancer is poor because of, the low chance of therapeutic treatment. The symptoms of HCC are usually absent. Further, the improvement of early detection markers is a vital missing component in methodologists to reduce HCC mortality.Chromosome aberrations in cancer cells can be resolved with a rising number of large-scale genomic and molecular genetics SNP array technology. However, the recent advancement of alteration of gene expression profiling and genome-wide copy number investigation have allowed the comprehensive selecting properties of whole cancer genomes. This finding enhances our comprehension of the epigenetic aberrations and genetic aberrations driving the development of liver cancer, instead of ultimately extend guidance to designed treatment of patients.SNP array is the most widely recognized strategy to examine the copy number of DNA sequences. It provides accurate information of CNAs which is essential to distinguish deletion and amplification in the chromosome. A better perception of gene expression in hepatic cancer can aid to comprehend the pathophysiology of HCC and enhanced prognostication.This work demonstrates that genomic high-density SNP-array test can be utilized to detect and characterize genome-wide CNAs on more than one thousand clones emerging from several chromosomes. By testing CBS and one-dimensional DSWT techniques on 10 HCC cell lines it has been found that there is a certain number of chromosome aberration including gains of 1q,8q,11q,17q,20q chromosomes and losses of 4q,8p,13q,16q,17p. A new altered region has been detected by amplification of chromosome 4 region 4q22.1. This gene expression profiling SSP1(secreted phosphoprotein 1) and AFF1(AF4/FMR2 family member 1) can prompt to: discover new biomarkers that may help enhance diagnostic for detecting HCC, represent novel objective for therapeutic agents, and allow us to identify regions in the HCC genome that have undergone recurrent high-level focal deletions or amplifications.