-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Clinical Medicine and Diagnostics
p-ISSN: 2163-1433 e-ISSN: 2163-1441
2015; 5(4): 60-69
doi:10.5923/j.cmd.20150504.02
Immunohistochemistry: A Revolutionary Technique in Laboratory Medicine
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLJude O. Okoye , Isaac N. Nnatuanya
Histopathology Unit, Department of Medical Laboratory Science, Madonna University, Elele Campus, Nigeria
Correspondence to: Jude O. Okoye , Histopathology Unit, Department of Medical Laboratory Science, Madonna University, Elele Campus, Nigeria.
| Email: |  |
Copyright © 2015 Scientific & Academic Publishing. All Rights Reserved.
Immunohistochemistry (IHC), also known as immunocytochemistry, refers to the process of detecting antigens (which may be protein or lipid) by exploiting the principle of antibodies binding specifically to antigens in histological and cytological preparations. IHC staining methods which could be by direct or indirect staining are based on immunofluorescence, immunoenzymologic staining or affinity histochemistry in relation to different biotin conjugated with antibodies. This technique which microscopically detects cellular constituents through antibodies of varying specificity has remodeled the domain of diagnostic pathology by hijacking the spotlight on the yet indispensible hematoxylin and eosin (H&E) technique. However, as with any other technique, the use of control samples is critical in accurate interpretation of IHC results for genetic and therapeutic procedures. The multi-staining, antigen retrieval and scoring techniques have further demonstrated the inestimable value of IHC in uncovering, classifying and/or identifying lesions which have earlier proved to be diagnostically elusive. Yet, more researches are still required in the area of its standardization for proper application in routine and clinical investigations in the face of overwhelming emerging diseases.
Keywords: Immunohistochemistry, Antigen Retrieval, Retriever Buffer, Scoring, Multi-Staining Technique, IHC Control, Application of IHC
Cite this paper: Jude O. Okoye , Isaac N. Nnatuanya , Immunohistochemistry: A Revolutionary Technique in Laboratory Medicine, Clinical Medicine and Diagnostics, Vol. 5 No. 4, 2015, pp. 60-69. doi: 10.5923/j.cmd.20150504.02.
Article Outline
1. Introduction
- Over the years, hematoxylin and eosin (H&E) technique have been considered the gold standard for the identification of pathologies in biological samples. However, quest for more knowledge in the pathogenesis and progression of some emerging diseases have questioned the indiscriminate continual usage of the H&E technique in the field of diagnostic pathology, hence the advent of Immunohistochemistry (IHC). Immunohistochemistry, a technique that microscopically detects cellular constituents via specific antibodies have been used for the detection and localization of a variety of microorganisms and other tissue proteins [1, 2]. Through comparative analysis, IHC has been discovered to advance the field of diagnostic pathology by significantly enhancing the diagnostic potency of the H&E slide like no other accessory technique in pathology. This has partially been facilitated by technical advances in immunohistochemistry including antigen retrieval methods, availability of sensitive detection systems, automated immunostainers, a broad range of antibodies applicable to routinely processed material, that is, surgical and cytologic material, all leading to high specificity and sensitivity [2]. IHC has played an eminent role in the histopathological classification of diseases. The detection of specific patterns of immunohistochemical expression and its likely association with a given diagnostic label has become a standard component of a pathologist’s diagnostic perspicacity and, in many ways, dictates much of the ground breaking research in diagnostic histopathology. The ultimate aim of introducing IHC to laboratory medicine is to achieve reproducible and consistent demonstration of antigens with the minimum of background staining whilst preserving the integrity of tissue architecture. This article reviews the principle, multiple-staining technique, antigen retrieval, interpretation and scoring of immunohistochemically stained tissues in modern medicine.
2. Sample Preparation
- Preparation of the sample is critical to maintain cell morphology, tissue architecture and the antigenecity of target epitopes. This requires proper tissue collection, fixation and sectioning. A solution of paraformaldehyde is often used to fix tissue, but other methods may be used. The tissue may then be sliced or used whole, dependent upon the purpose of the experiment or the tissue itself. Before sectioning, the tissue sample may be embedded in a medium, like paraffin wax or cryomedia. Section can be sliced on a variety of instruments, most commonly a microtome or cryostat, and are sliced at a range of 4-40μm. The slices are then mounted on slides, dehydrated using alcohol washes of increasing concentrations (e.g., 70 %, 90 %, 95 %, and 100 %) and cleared in xylene before being imaged under a microscope. Dependent on the tissue type and the method of antigen detection, endogenous biotin or enzymes may need to be blocked or quenched, respectively, prior to antibody staining. Although antibodies shows preferential avidity for specific epitopes, they may partially or weakly bind to sites on nonspecific proteins (also called reactive sites) that are similar to the cognate binding sites on the target antigen. A great amount of non-specific binding causes high background staining which will mask the detection of the largest antigen. To reduce background staining in IHC, and other immunostaining methods, samples are incubated with a buffer that blocks the reactive sites to which the primary or secondary antibodies may otherwise bind. Common blocking buffers include normal serum, non-fat dry milk, BSA (Bovine Serum Albumin) or gelatin. Methods to eliminate background staining include dilution of the primary or secondary antibodies, changing the time or temperature of incubation and using a different detection system or different primary antibody [3].
2.1. Antigen Retrieval (AR)
- Tissue that has been subjected to fixation with formalin shows a partial or complete loss of immunoreactivity due to epitope masking. Formaldehyde covalently binds to tissue protein and also acts to cross link adjacent proteins or peptides to form large aggregates of proteins. Intra and inter molecular links introduced by formaldehyde fixation alters the protein secondary, tertiary and quaternary structures, thereby lowering the accessibility of the epitope sites. The active ingredient in formalin is formaldehyde. So, although formalin fixation preserves tissue morphology, it also alters the three dimensional structure of the tissue proteins. This alteration can lead to a modification of the antigen’s epitopes. This modification results in an antigen’s inability to react with the paratope of an antibody and can only be corrected by restoration of the epitope also known as antigen retrieval or demasking of antigens (figure 1). Antigen retrieval refers to any technique in which the masking of an epitope is reversed and epitope-antibody binding is restored. The need for antigen retrieval depends on multiple variables such as the duration of fixation, type of antibody used (monoclonal vs. polyclonal). Monoclonal can be mouse or rabbit hybridoma, tends to be ‘cleaner’, very consistent batch-to-batch and are more likely to get false negative results Polyclonal are of many different species, tend to have more non-specific reactivity, can have very different avidity/affinity batch-to-batch and are more likely to have success in an unknown application. Recent advances in antigen unmasking techniques have provided better results and include: digestion with proteolytic enzyme, heat and or pressure treatment, and use of specific fixatives [2].
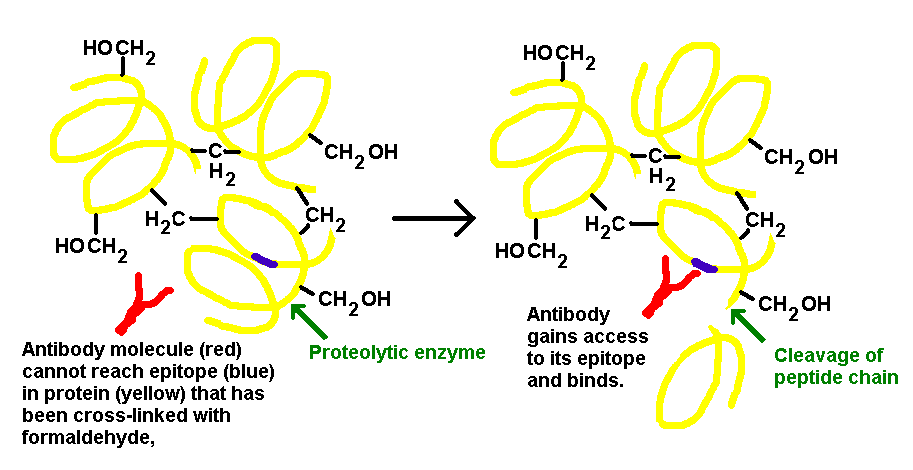 | Figure 1. Principle of proteolytic enzyme action in Unmasking of an Epitope |
2.2. Retrieval Buffers
- Since the development of HIER, a wide range of buffered solutions have been employed, it is the responsibility of each individual laboratory to determine which AR solution will perform optimally for each antigen/antibody, pH is very important in maintaining an optimal morphology and consistent immunoreactivity. HIER solutions can be grouped into 3 categories based on pH and buffer comparisons: Low pH (pH approximately 3-5) solutions buffered by glycine-HCl, Low to neutral pH (pH approximately 6-7) solution buffered with citric acid, High pH (pH approx. 8-10) buffered by Tris or EDTA. High pH Buffers (EDTA) pH 8-10 are effective on over fixed specimens and tend to increase staining intensity of most antibodies while its disadvantages more likely to include tissue loss and distortion of tissue morphology which makes the nuclei appear convoluted and bizarre. Low pH buffers (Citrate) pH 6-7 tends to preserve tissue morphology and demonstrates a distinct nuclear pattern staining intensity. Their disadvantage or limitation is that they can be ineffective on over fixed tissue.
3. Multiple-Staining in Immunohistochemistry
- Multiple staining can be defined as the detection of two or more targets on one slide, thus increasing the information obtained from each slide and reducing turnaround time, compared to single staining or sequential staining. Multiple staining, like single staining, can be performed on any of Formalin Fixed Paraffin Embedded (FFPE) tissue sections, frozen sections, cell smears and cytospin preparations. This technique also makes it possible to assess the topographic relationship of two or more targets, for example, to determine whether targets are present in different cell populations, in different cells, in the same cell, or even in the same cellular compartment. In addition, multiple staining allows the combination of in situ hybridization (ISH) and IHC, giving information about a particular target both at protein level and DNA/mRNA level. Information can also be obtained on possible cell-to-cell spatial contacts of different cell types. Furthermore, with an increasing demand for less invasive sampling techniques and smaller and fewer specimens available, multiple staining has the advantage not only of conserving tissue, but also saving time and reagents.The diagnosis of prostatic intra-epithelial neoplasia (PIN) is just one example of the clinical importance of multiple staining. Prostate needle biopsy is the preferred method for diagnosing early prostate cancer, but in some cases the diagnosis is uncertain because the biopsy includes only a few malignant glands, or a few hyperplastic or dysplastic glands that are difficult to distinguish from cancer [4, 5]. Since basal cells typically are present in hyperplastic, and dysplastic glands, as well as around ‘in situ’ (PIN) lesions, but absent in malignant invasive glands, the demonstration of basal cells can be used to assist recognition, or exclusion, of invasive cancer. Basal cells are labeled using high molecular weight cytokeratin, cytokeratin (e.g. CK5/6 - cytoplasmic) or p63 (nuclear) immunostaining, or both. In addition, AMACR/P504S, is expressed in a high percentage of prostate carcinomas, but is negative or only weakly expressed in benign prostate tissue. Thus it is used as a positive cancer marker, often in a multiplex stain with keratin and p63. If single stains are done on serial sections, interpretation is much more difficult and ambiguous lesions may be absent in adjacent cuts, especially when dealing with small foci, with the result that some malignancies may remain undiagnosed. In this context, multiple staining protocols significantly improve the ability to distinguish between benign and malignant lesions. This approach, which reduces the percentage of ambiguous lesions and the need for additional biopsies, is being extended to facilitate recognition of other invasive cancers, as in breast.
3.1. Technical Challenges
- Most primary antibodies used today originate from either mouse or rabbit and are visualized using systems based on anti-mouse and anti-rabbit secondary antibodies. The challenge of distinguishing between two primary antibodies of the same species (mouse-mouse, or rabbit-rabbit) must be addressed, because separate mouse and rabbit primary antibodies to the chosen targets often are not available. Utilizing two primary antibodies of the same species can require quite elaborate protocols. Spectral differentiations of stain colors may be difficult; especially if the targets are co-localized leading to a mixture of colors [6]. The ‘mixed’ color should contrast well with the two basic colors. In the case where a rare target is co-localized, the color reaction of the more abundant target will tend to dominate the other. Even if targets are not co-localized it is difficult to balance signals so as to enable visualization of a rare target in the same slide as highly expressed targets. The use of Image analysis approach (spectral separation) or an adjustment in concentration of the primary antibodies may solve this problem.
3.2. Multi-Staining Method Selection
- To ensure success, IHC staining using multiple antibodies must be carefully planned. If primary antibodies of the desired specificity for the two (or more) targets are commercially available, and made in different species, then there are several different staining methods that one can choose. However, very often the choice may be limited by the reagents available [7]. Care must be taken to avoid cross-reactivity between reagents; in the event that avoidance is not possible, then measures must be taken to minimize the risk, including additional controls to detect significant cross reactivity if present. In general, staining methods can be divided into the following classes:
3.2.1. Sequential Staining
- By this method, one staining procedure succeeds another. For example, the first antibody is applied to the tissue section followed by a labeled detection system such as streptavidin-biotin horseradish peroxidase (HRP), with a chromogen such as DAB. The second primary antibody is applied only after the excess DAB is rinsed off, followed by labeling with a streptavidin-biotin alkaline phosphatase (AP) detection system and a colored chromogen. The biggest advantage of sequential staining is that by this procedure problems related to cross-reactivity are minimized, possibly due to steric interference. The disadvantages of sequential staining are: the method cannot be used for co-localized targets, the technique often leads to a long staining protocol and carries an inherent risk of incorrect double staining due to incomplete elution of unreacted reagents from the first staining sequence, before application of the next reagents. Elution may become an issue with some high-affinity primary antibodies, as these may remain at their binding-site, leading to spurious double stained structures. Elution also risks denaturing epitopes of antigens to be visualized subsequently. Furthermore, for some chromogens there is a risk that the first chromogen (DAB in particular) may shield other targets. This technique is, therefore, not recommended for evaluation of mixed colors at sites of co-localization, because not all reaction products are capable of surviving the rigorous washing required to remove the antibodies. To avoid such problems and blurry staining results, it is recommended to use the most ‘robust’ dyes such as DAB, Fast Red, AEC and Blue chromogen first, followed by other less ‘robust’ dyes.
3.2.2. Simultaneous Staining
- In a simultaneous double stain, the primary antibodies can be applied simultaneously. The advantage of this method is that it is less time-consuming because the reagents can be mixed together. However, the technique can only be used if the primary antibodies are from different species, or are directly labeled with different enzymes [8].
4. Interpretation of Immunostains
- Most irregularities among published reports of Immunohistochemical assays are readily attributable to procedural deviation, and many conflicting results in the literature are the result of technical reasons, particularly variations in the choice of fixatives and duration of fixation. Thus, standardization of tissue processing and immunostaining across laboratories would be a useful initial step. Efforts to achieve such standardized procedures have been made [9-11]. In recent times, whether epitope retrieval was used, and by which means, has added another source of conflict. However, many discrepancies are the result of variance in the interpretation of the immunostains or to difficulties in communicating the findings, including criteria for what is considered to be a positive staining. These problems are well recognized by most pathologists, although there are only occasional attempts in the literature to design more general standards for the interpretation of Immunohistochemistry [10, 12, 13]. The interpretation of many Immunohistochemical stains is qualitative and subjective, with quantification of the reaction having little or no importance. Often a diagnostic decision is based on whether a certain molecule is expressed or not by cells. Thus, any amount of detectable leukocyte common antigen, for example, is often enough to tilt the diagnosis in favor of lymphoma, given the appropriate context of differential diagnosis. Proper interpretation of most immunostains, however, depends to some extent on estimation of antigen content and in the establishment of cutoff levels between positive and negative results. Reasonable reproducibility, from run to run, is essential for these cutoff levels to work. Additionally, threshold levels require adjustment to methodology, in particular to method sensitivity. For all the above reasons, achieving reproducible interlaboratory thresholds is one of the most difficult challenges we face today. As quantitative tissue-based biochemical assays are progressively replaced by immunohistochemistry, interest in accurately measuring immunostains has increased. Some may question whether such precision is currently attainable, or even necessary. Numerous methods for visual scoring of Immunohistochemical assays have been proposed to improve quan-titation. These have been shown to be better reproducible than visual estimates and often to be of clinical relevance. However, these scoring systems suffer from their own interobserver reproducibility problems [14]. Computer-assisted image analysis has proven superior to visual estimates in providing quantitative Immunohistochemical assays, particularly when applied to frozen sections or fine needle aspirations [15]. These systems, particularly if automated, hold the promise of improving the accuracy and reproducibility of quantitative Immunohistochemistry as well as becoming tools for more accurate interlaboratory and intralaboratory quality control.Causes of discrepancy in the interpretation of immunostains are numerous. An increasingly common cause of contradictory reports nowadays is the lack of well-defined standards about what constitutes a positive result. For example, most investigators require cytoplasmic and nuclear expression to interpret S-100 protein immunostains as positive. However, there are still a few who would interpret, as positive, cases in which only cytoplasmic staining is present. Because heterologous (usually rabbit) antisera are predominantly used for this assay, it is safe to assume that many such cases showing only cytoplasmic staining may be spurious. Similarly, some early immunohistochemical studies of the HER-2/neu protein included, as positive, cases in which cytoplasmic staining was present [16-18]. It is known that only cases with cell membrane staining are associated with amplification of this gene as determined by molecular methods of detection. A simple rule to prevent some of these problems is that, when the location of the target molecule is known, the pattern of immunoreactivity must follow the microanatomic distribution of the antigen. For example, a granular intracytoplasmic pattern should be seen with antibodies that detect cell products packaged within cytoplasmic granules (chromogranins, von Willebrand factor, HMB-45, etc.). Immunohistochemical detection of cytokeratins should result in a finely fibrillary intracytoplasmic pattern. Thus, when any stain does not conform to these criteria, a falsely positive stain should be suspected. Thin, well-stained sections using high-resolution chromogens are essential for this rule to work. In our experience diaminobenzidine hydrochloride offers the best resolution to date. Validation of results, in cases of ambiguity, should be sought by using antibodies to different epitopes of the same molecule, or detection of related markers (for example, synaptophysin and chromogranin are often expressed together by neuroendocrine tumors). However, whenever the nature of the antigen is not well characterized, determining what constitutes a positive result is more difficult to ascertain. For example, according to some authors, the antibody BerEp4 stains many adenocarcinomas and rarely, if ever, stains mesotheliomas [19, 20]. Others, using similar methods and the same antibody, have reported as many as 10% of mesotheliomas to stain with BerEp4 [21]. Latza et al. [22] in their initial publication on BerEp4, stated that its immunoreactivity was predominantly located at the basolateral cytoplasmic membranes of epithelial cell. It appears that some discrepancies in the literature stem from whether the authors choose to interpret apical membrane staining as positive or not [20]. Another source of problems is that a quantitative approach to distinguish positive from negative immunostains is sometimes used by authors. Often the author’s cutoff levels are not easily exported to other laboratories and even their intralaboratory reproducibility has not been tested. Unfortunately, at the present time, there is no consensus as to what constitutes an adequate threshold of interpretation for most immunostains. For example, some laboratories include, as positive estrogen receptor, any case in which even a single tumor cell shows any degree of detectable reactivity. Others require at least 20% of the cells to immunoreact. To make things worse, often these cutoffs have been arbitrarily set without the validation of a clinicopathologic study. It has been suggested that laboratories that have not conducted clinical validation studies adhere to the methods and interpretation criteria of those laboratories that have successfully done so [23], a view we endorse. However, it is important to keep in perspective that when a retrospective study is carried out for such clinicopathologic validation, it is usually done on selected case material from a single institution, with relatively uniform fixation and processing. Moreover, to minimize daily variation, slides are immunostained together in a single run [24]. A cutoff level is then sought, usually with aid of statistical analysis. This procedure is perfectly reasonable to minimize variations because of method and is particularly applicable when comparing different antibodies or methods. Unfortunately, this approach has little bearing on real life immunohistochemistry. Unpublished studies (H.B.), for example, have shown daily variation in optical density of as much as 30% in immunostains for estrogen receptor, when the same block of tissue was used as a daily control. Clearly, the variation was methodologic, despite the fact that an automated processor was used. Add to this divergence of fixation and processing, as in the case of specimens handled by a reference laboratory, and the complexity of the problem comes into focus. Evidently, there is need to establish uniformity in the setting of thresholds.
5. Quantitative Immunohistochemistry
- Whether the ultimate goal is to find clinically meaningful cutoff levels or the accurate measurement of antigen molecules per cell, interlaboratory reproducibility of immunostains is fundamental. The use of improved control methods, as discussed below, is also essential. In addition to commonly used semi-quantitative estimations of immunohistochemical stainings, using arbitrary scoring systems, at least two separate forms of quantitative immunohistochemistry are readily evident. In its simplest form merely events are measured, with no attempt to assay for the quantity of analyte expressed. Examples of this type are counting micrometastases in bone marrow samples or measuring peritumoral blood vessels. In these examples, minor variations in the intensity of the immunoreactivity, attributable to the method of staining or fixation procedure, have little impact on the quantification itself. With good control of specimen preparation and staining procedures, these quantitative procedures are currently attainable by most laboratories. A similar quantitative approach can be exemplified by the estimation of proliferation index with staining for Ki-67 (MIB-1) where a simple count can be performed on a given amount of normal or neoplastic cells. Methods like these allow quantitative estimation but may not be highly reliable and reproducible in terms of a specified cutoff value. Accurate conversion of immunoreactivity into levels of analyte per sample is much more complex, requires special equipment, and given the vagaries of fixation and processing, is not currently possible on archival paraffin embedded material. Several considerations have been advocated for scoring in Immunohistochemistry, which include:Considerations for scoring positivity: Semi-quantitative method, Intensity of staining – none, weak, moderate, strong, Proportion of tissue stained in % - <5-10%, 10-25%, 25-50%, 50-75 %, >75% and Combining the scores to give an overall index.Considerations for scoring negativity includes: when the Ag is not present or, Not present in a significant amount (<5-10%) or Nothing more than the normal expected e.g. mitosis marker Ki-67.Factors resulting in false positivity includes: cross reactivity of substances, Non-specific binding of antibodies to antigen, Presence of endogenous products, Entrapment of reagents by normal tissues.Factors resulting in false negative results includes: inappropriate, denatured, wrong concentration.
6. Controls in Quantitative Immunohistochemistry
- Sections of tissue expressing the target molecule and tissues known not to express it are currently used routinely in immunohistochemical procedures in virtually every laboratory. Figure 2a is an H&E stained control section while figure 2b and 2c serve as immunopositive and immunonegative control slides, respectively for breast cancer diagnosis. The limitation with such latter control tissues is that, at best, they only serve to control the immunostaining procedure itself. Other sources of variation such as fixation and processing are not controlled. Moreover, the amount of target molecule present in the control tissues is, more often than not, unknown. The ideal control for quantitative Immunohistochemistry should apply to the entire procedure, from fixation to interpretation of results. A suggested possible approach is to suspend in a solid matrix, cultured cells, expressing known and independently measured quantities of the target molecule and to place such artificial tissues within the tissue cassette alongside the specimen (Quicgel) [25]. By this means both specimen and control are simultaneously subjected to fixation, processing, antigen retrieval, staining, and interpretation. However, such a procedure does not eliminate differences as a result of antigen degradation during transport of fresh specimens or fixation time if the specimens are delivered fixed, which may be of importance in certain situations. This would be particularly useful in cases in which the need for quantitative immunohistochemical assays is anticipated, as in the case of breast biopsies. Some progress along these lines has been recently reported using breast cancer cell lines that express a known amount of hormone receptors [26].
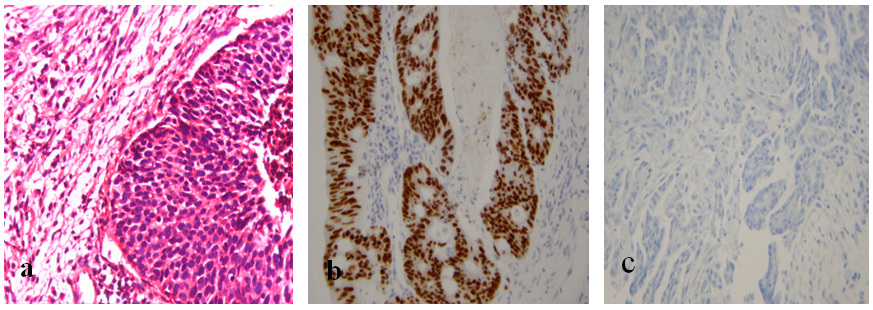 | Figure 2. A-photomicrograph of a malignant breast tissue section stained by H&E technique (x400), B-photomicrograph of an immunopositive (LM 28) breast tissue section showing nuclear staining (marked by brown staining dots, x400) and C-photomicrograph of immunonegative breast tissue section (x400) |
7. Application of IHC
- IHC can be used to demonstrate the earliest changes in transformed tissues, identifying cellular changes not normally visible with H&E. Individual markers for proliferation, apoptosis and specific tumor proteins can be used to help distinguish hyperplasia from neoplasia and determines specific tumor origin/type. IHC provides a relatively rapid and simple method to better determine the origin of neoplastic tissue or investigate the behavior or progression of a given neoplasm. The three distinct roles of IHC which include: (1) diagnostic IHC; (2) genetic IHC and (3) therapeutic IHC have their individual impacts on modern diagnostic pathology, and are further discussed below.
7.1. The Diagnostic Application of IHC
- The application of IHC can provide valuable diagnostic information in the initial determination of malignancy. Specific antibodies directed against a range of generic tumour markers determine their presence/absence in disease tissue versus normal, as well as the specific diagnostic label for a given set of histological changes. Although the IHC analysis is often interpretative and, thus, carries a reduced specificity, the identification of immunoreactivity patterns can confirm tumour type. Furthermore, when utilised in combinations (so-called immunopanels, discussed later) and interpreted in the correct clinical context, the value of such markers is greatly enhanced. The classic example of generic tumour markers is presented by the Cytokeratins (CKs). These are epithelial markers useful for confirming the epithelial nature of tumours and, hence, designation as carcinoma [27, 28]. Normally, the expression of CKs varies with epithelial cell type, extent of differentiation and tissue development [29]; however, during malignant transformation, the CK patterns and integrity are maintained, a property that enables their use as tumour markers [30]. Unfortunately, very few CK markers are organ-specific which limits their utility. This non-specificity is a feature of many commonly used antibodies. Smooth Muscle Actin IHC, which identifies tissue of smooth muscle or myofibroblastic origin, can be positive in many non-muscular / myofibroblastic lesions. Additionally, reactive myofibroblasts are present in several tumour types from various origins [28]. CD34 is immunoreactive in many soft tissue tumours, including vascular, solitary fibrous, gastrointestinal stromal, peripheral nerve sheath, epithelioid sarcomas and in a subpopulation of dermal dendritic cells. Due to its non-specificity, this marker is usually complemented by additional markers, such as CD31 for the diagnosis of vascular tumours and CD117 (KIT) or DOG1 for gastrointestinal tumours [28]. Interestingly, it would appear that in the context of diagnostic IHC, the results always provide a diagnostic likelihood but not a diagnostic certainty, hence, the interpretative approach and the need to consider morphology and wider clinical context. Due to the mentioned lack of specificity of these single generic tumour markers, diagnostic immunopanels comprising several immunohistochemical stains are employed. Such an approach increases diagnostic accuracy and strengthens single biomarker evaluation. Several examples of diagnostic immunopanels exist. The lymphoma panel is broadly based on morphologic differential diagnosis, and derived from knowledge of lymphocyte development and anatomic compartmentalization within the lymph node: expression of markers associated with specific stages of lymphocyte development and the immunoarchitectural features facilitate diagnosis. The precise immunopanel varies with the initial morphological analysis and likely differential diagnosis. However, it usually includes analysis of CD20, PAX5 (B cell) and CD3, CD4, CD5, CD8 (T cell) expression. Anatomic architectural alterations are also evaluated: BCL-2, CD10 (follicular patterns) and other markers include CD45, CD23, cyclinD1, CD15 and CD30. Additional markers can identify subgroups of lymphoma [31, 32]. For example; BCL-2 and CD10 characterize the germinal-centre phenotype, typified by follicular lymphoma while post-germinal-centre lymphomas are in the plasma cell pathway and correspondingly express CD138 and MUM1. Although beneficial as a tumour diagnostic, this panel-based approach holds limited genetic or therapeutic value [33].The ability to immunohistochemically demonstrate any antigenic substance that is at least partially retained in tissues coupled with the advent of techniques generating monoclonal antibodies, has led to the availability of a myriad collection of new antibodies for the pathologist and scientist. They vary from a few that are highly tumor specific to others that demonstrate low antigen expression in normal cells and high expression in malignancies. Most show antigen expression directly related to the differentiation of the tumor such that the sensitivity of these markers is highest in well-differentiated neoplasms and low to undifferentiated in poorly differentiated ones. Thus, it is important not to interpret negative staining solely as such, but also to recognize it as possibly representative of a poorly differentiated neoplasm lacking antigenic expression or showing expression so minimal as to be undetectable. Even though many antigens are tissue specific, there may be considerable overlap with other neoplasms. In addition, the microanatomic distribution (i.e., nuclear, membranous, or cytoplasmic) of some antigens may be different in tumors and normal tissues, or even among different neoplasms. For example, carcinoembryonic antigen (CEA) has an apical or luminal distribution in normal cells but a more random cytoplasmic pattern in malignant cells. Secondly, in contrast to monoclonal CEA that shows cytoplasmic reactivity, polyclonal CEA shows a canalicular pattern of staining in hepatocellular carcinoma. Another example is CK20 that demonstrates membranous staining in many normal and malignant epithelial cells; with the exception of Merkel cell carcinoma in which it shows perinuclear dot-like staining [2].In the workup of the metastatic carcinoma of unknown origin, the plethora of Immunohistochemical markers should be applied in an algorithmic fashion, based on a logical succession of interpretive steps. This approach enables one to increase the specificity of the antibodies that are nonspecific by coupling them with others that are lineage specific. Initially, broad spectrum antibodies such as Cytokeratin (CK), Vimentin, S100 protein, and Leukocyte common antigen should be used to widely categorize neoplasms as follows: carcinomas, lymphomas, sarcomas, and melanomas [2]. A more selective panel of IHC stains based on the clinical history and radiologic impression may then follow. Despite the fact that the use of immunohistochemistry has been found to have a cost-effective advantage in the evaluation of the unknown primary over other clinical tests, one must discriminate wisely in selecting the economically favorable immunohistochemical panel. Prior to discussing the array of antibodies for tumor classification, it is important to realize that Immunohistochemistry is an ancillary tool, and thus, must be interpreted in the context of a detailed morphological analysis and well-formulated differential diagnosis based on the H&E [2]. Some normal and malignant cells which have been successfully classified using IHC are as follows:
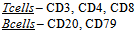 DLBCL – CD20Mantle cell lymphoma – Cyclin D1Marginal zone lymphoma – CD10Follicular lymphoma – BCL-2Hodgkins lymphoma – CD15, CD30, CD20Small cell lymphoma (CLL) – CD5, CD23
DLBCL – CD20Mantle cell lymphoma – Cyclin D1Marginal zone lymphoma – CD10Follicular lymphoma – BCL-2Hodgkins lymphoma – CD15, CD30, CD20Small cell lymphoma (CLL) – CD5, CD237.2. The Genetic Application of IHC
- In this particular case, the gain or loss of protein expression detected by an IHC becomes a surrogate of an inherited mutation. Many examples exist whereby the mutational status of certain biomarkers dictates the overexpression/diminished expression of the resultant proteins. Furthermore, IHC analysis of this nature can account for the genetic variability of individuals within the same population, that is, the same cancer type. Mismatch repair (MMR) gene mutations are the most characterized forms of genetic instability in colorectal cancer (CRC) and hereditary non-polyposis colorectal cancer (HNPCC)/Lynch syndrome. Microsatellite instability (MSI) is a hallmark of HNPCC [34]. The persistence of mismatch mutations as a result of defective MMR proteins and enhanced MSI predisposes individuals to HNPCC and CRC [35]. HNPCC sufferers inherit one germline mutation in an MMR gene. In this instance, the complete loss or patchy/weak expression of the MMR IHC (MSH2, MSH6 and MLH1, PMS2) has important clinical implications indicating either absent MMR protein, expression of a truncated protein, or loss of the epitope recognised by the antibody due to mutation. It is noteworthy that a small proportion of HNPCC-related tumours do not exhibit abnormal MMR protein expression by IHC, even though the function of the MMR system is defective [36]. Indeed, the decision of what should be analyzed first (MMR IHC or MSI status) in the overall diagnosis of HNPCC and its cost-effectiveness is an old debate in diagnostic laboratories [37] and the choice is usually dictated by the availability of one of those methods in non-integrated laboratory environments.
7.3. The Therapeutic Application of IHC
- The therapeutic information gained from specific biomarker expression tissue studies reaffirms the multifaceted role of IHC in cancer. Assessment of these biomarkers can be directly aligned with specific treatment options for individuals as well as providing important prognostic and predictive information. Relying on near-to-absolute quantification, IHC scores can inform likelihood of response to targeted treatment. It is well established that certain tumors originating in reproductive organs, such as the breast, prostate, endometrium, and ovary, are partially regulated by hormones. The growth regulation of these tumors is mediated through expression of specific receptors for hormones so that tumors expressing high receptor levels respond to hormone ablation therapy and vice versa. In breast cancer, the determination of hormone receptor status is therapeutically important and is routinely done to guide hormonal therapy. This is facilitated by monoclonal antibodies that recognize epitopes of the receptor proteins on paraffin-embedded tissue, a method superior to using frozen tissue and cytosol-based methods [38, 39]. In addition, it permits quantitative and qualitative assessment even on small tissue specimens such as core biopsies and aspirate material. Expression correlates directly with response to hormonal treatment and inversely with factors such as tumor grade, ploidy, and stage. Two multidrug resistants (MDR) genes (1 and 3) have been described, but only 1 [40], confers the MDR phenotype. P-glycoprotein (P-170), a transmembrane protein, is responsible for most cases of multidrug resistance. Expression correlates with resistance to chemotherapeutic agents such as anthracyclines and vinca alkaloids. It functions by operating an energy dependent pump leading to a decrease in the intracellular accumulation of the drug and resulting in an MDR phenotype. Thus, tumors responsive to chemotherapy express little to no P-glycoprotein and vice versa. Immunohistochemical detection of P-glycoprotein is useful since it identifies tumors that may be responsive or resistant to chemotherapy.
8. Conclusions
- Immunohistochemistry (IHC) combines histological, immunological and biochemical techniques for the identification of specific tissue components following the principle of specific antigen/antibody reaction tagged with a visible label. Its use in the study of cellular markers that define specific phenotypes has provided important diagnostic, prognostic and predictive information relative to disease status and biology. The multi-staining technique involved and possibility of antigen retrieval has made IHC a revolutionary tool in laboratory medicine. However, the key to IHC universal acceptance and maximum utilization requires more research on its standardization in relation to scoring of stained tissues and interpretation of result.
ACKNOWLEDGEMENTS
- Author wishes to acknowledge the efforts of Kemi Erhiaganoma (CTO Gemrook Diagnostic Services, Nigeria) and Olorunda Rotimi (St. James’s University Hospital, UK) in introducing the concept of Immunohistochemistry to several research centres in Nigeria and their contributions to this review paper.