-
Paper Information
- Next Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
Clinical Medicine and Diagnostics
p-ISSN: 2163-1433 e-ISSN: 2163-1441
2011; 1(1): 1-7
doi: 10.5923/j.cmd.20110101.01
Central Sensitization: Clinical Implications for Chronic Head and Neck Pain
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-Text HTML
Full-Text HTMLArthur S. Roberts
Department of Oral Medicine, Indiana University School of Dentistry, 46202, Indianapolis
Correspondence to: Arthur S. Roberts , Department of Oral Medicine, Indiana University School of Dentistry, 46202, Indianapolis.
| Email: |  |
Copyright © 2012 Scientific & Academic Publishing. All Rights Reserved.
Chronic clinical pain associated with CS, is a potentially progressive, devastating, multimodal disease with a significant worldwide economic and social burden. Effective intervention is dependent upon recognizing the fundamental differences in acute and chronic pain, the effects on and by the neuromatrix upon the biopsychosocial health of the individual patient, and integrating that knowledge into a comprehensive multidisciplinary therapeutic plan.
Keywords: Chronic Pain, Sensitization, Neuromatrix, Biopsychosocial, Polypharmacy
Cite this paper: Arthur S. Roberts , "Central Sensitization: Clinical Implications for Chronic Head and Neck Pain", Clinical Medicine and Diagnostics, Vol. 1 No. 1, 2011, pp. 1-7. doi: 10.5923/j.cmd.20110101.01.
Article Outline
1. Introduction
- Chronic pain is a multidimensional experience encompassing biological, psychological and social elements. Each of these elements is entangled bi-directionally with the neuromatrix as a whole. Figure 1.There is a growing consensus that chronic pain is a disease entity. This disease has been termed maldynia by the American Medical Association. This new designation has come about in recognition that acute and chronic pain are fundamentally different experiences, demanding a fresh approach to diagnosis, therapy, and prognosis. It has been repeatedly demonstrated that many chronic pain disorders have a major component of central sensitization. Assessment and therapy for these several disorders is dependent on an understanding of central sensitization.
2. What is Central Sensitization (CS)
- Central sensitization is thought to occur when “Sensory-afferent signals overwhelm the body's ability to filter them”.[1,2] When this occurs a number of pathophysiologic changes occur including neuro-immune dysfunction, neuro-endodrine dysfunction, NMDA (N-methyl-D-aspartate) dysregulation, sympatho-afferent coupling; and altered serotonin and norepinephrine production and utilization. These events are thought to occur predominantly in the mid-brain and associated structures and are influenced by elements of the neuromatrix delineated by Melzack, Woolf, and others.[1,3-5]These pathophysiologic changes are associated with decr-eased descending inhibition, dysautonomia, and altered serotonin production/utilzation. The resulting depression, anxiety, sleep fragmentation, allodynia, and hyperalgesia characterize a number of chronic pain disorders.[2-13]At least two etiological pathways are posited for central sensitization. The first is chronification of nociceptive pain; and involves neuroplastic changes, and peripheral sensitization as precursors to central sensitization and the resulting clinical pain. A more psychologically centered pathway is offered as an alternative to this transformed nociception. This alternate pathway begins with elevated levels of chronic stress, and encompasses elements of anxiety, sleep fragmentation, decreased pain thresholds, and dysautonomia. It is common to find both axes actively involved in the etiology of a central sensitization syndrome. Central sensitization syndromes can be thought of as the archetypical exemplars of a chronic biopsychosocial pain disorder.[1,3,5,14-18].
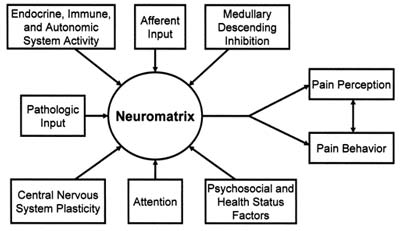 | Figure 1. |
2.1. What Are Central Sensitization Syndromes (CSS)
- “A similar and overlapping group of syndromes without demonstrable pathology and bound by a common patho-physilogical mechanism of central sensitization.”[2] The balance of expert opinion holds that the following are part of that group: Migraine headache, tension-type headache, chronic fatigue syndrome, fibromyalgia, burning mouth syndrome, atypical odontalgia, tempormandibular disorder, and myofascial pain syndrome. In addition, there is a growing consensus that multiple chemical sensitivity, post traumatic stress disorder, depression, primary dysmenorrhea, irritable bowel syndrome, periodic limb movement disorder and restless leg syndrome are strong candidates for inclusion as central sensitization syndromes. [2, 17, 19] Figure 2.
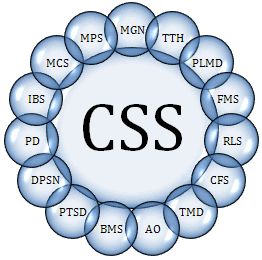 | Figure 2. Adapted from Wallace and Clauw [2] MGN – Migraine, TTH - Tension-type headache, IBS - Irritable bowel syndrome, CFS - Chronic Fatigue syndrome, FMS – Fibromyalgia, MPS – Myofascial pain syndrome, TMD – Tempoomandibulal dysfunction, RLS - Restless leg syndrome, PLMD - Periodic limb movement disorder, MCS - Multiple chemical sensitivity, PTSD - Post traumatic stress disorder, DPSN – Depression, PD - Primary dysmenorrhea, AO - Atypical odontalgia, BMS – Burning mouth syndrome. |
3. Diagnosis, Therapy and Prognosis
- By definition CSS are without demonstrable pathology. This leads us to clinical assessment as the most productive diagnostic approach. Allodynia, but not hyperalgesia, is an indicator of the presence of CS, as is exacerbated widespread pain highly correlated with elevated stress levels. The concurrent presentation of multiple disorders with a significant CS component is an indicator of the presence of central sensitization syndrome. [1-3]More specifically we can assess the patient to establish the presence of characteristic sequelae of the pathophsiologic changes associated with central sensitization. These are posited to be: Ÿ Vagal dysregulation reduces endorphin release and alters serotonin production and utilization. This produces altered accommodation of minimally painful events, and contributes to depression. [7, 18, 20]Ÿ Sympatho-afferent coupling of sensitized trigeminal complex leads to lowered parasympathetic drive and increased sympathetic drive. This contributes to dysfunctional sleep, and anxiety via increased norepinephrine levels.[6,21-25]Ÿ Decreased medullary descending inhibition of nociception, This increases the effective peripheral nociceptive input, and is reflected in lowered pain thresholds, hyperalgesia, allodynia, and greater impact of peripheral sensitization.[8,11,12,15-17,23,26-32]Ÿ Hypoactivity of the hypothalamic-pituitary-adrenal axis, and autonomic nervous system alterations, produce increased sympathetic tone, low vagal tone, and contribute to immune abnormalities. This contributes to the fatigue and malaise often associated with CSS. [5,12,14,20,21,23-25,33-38]Utilizing the resultant depression, anxiety, hyperalgesia, allodynia, stress related pain exacerbation, fatigue, and poor sleep, we can establish a list of indicators for central sensitization. Taken alone, with the possible exception of allodynia and stress related pain, none of these are pathognomonic. However, in concert they are highly suggestive of CS. In addition, when seen in association with other disorders thought to have a significant CS component, the probability of a CSS diagnosis is further increased.[1-3,5,13,14,39]Stress, a major contributing etiologic factor in CS can be assessed using a number of non-invasive methodologies: Ÿ Autonomic nervous system – salivary alpha-amylase and heart rate variabilityŸ Neural and immune profiles – cutaneous sweat patch
3.1. Therapy
- Polypharmacy is one of the problems attendant to CSS therapy, and is the result of approaching each of the varied presentations of CSS as a separate and distinct disease. Each complaint not intuitively associated with CS, by either patient or provider, may result in treatment by a number of different prescribing specialists. This inevitably leads to polypharmacy, and iatrogenic contribution to decreased patient quality of life, secondary to the side effects of the multiple medications. A part of this issue may derive from failing to differentiate chronic from acute pain. While the acute symptoms of CSS disorders need to be addressed, it is essential to treat the pathways in chronic pain disease. The pathways are few, and have fairly specific therapeutic protocols associated with them. [2,5,7,8,13-15,17,19,20,32]There are two distinctly different approaches to CSS therapy. The first, and historically more common, is to address the effects of CS after it has occurred. The second is to interrupt the CS and let the body’s homeostatic mechanisms clear residual pathologic products. Optimal outcomes often depend on doing both. Within these two broad categories we have both pharmacological and non-pharmacological therapeutic options.[1,4,6,8,13,14, 40-44]
3.1.1. Pharmacological Approaches
- Central sensitization is entangled with neuroplasticity and peripheral sensitization in many chronic pain syndromes, especially those deriving from transformed nociceptive pain.[1,2,13] Use of anti-inflammatory drugs may reduce levels of peripheral stimulus, but do not directly treat the central sensitization.[13]Many of the drugs that we use to address central sensitization will have a beneficial effect on the ability of the patient to cope with the pain, as well as to diminish the central sensitization itself.[1,2,8,9,13,41,45]Drugs used to address the effects of central sensitization:Ÿ AcetaminophenŸ Serotonin (SSRI) and norepinephrine (SNRI) reuptake inhibitors and tricyclic antidepressants (TCA)Ÿ Opioids and TramadolDrugs that may treat the central sensitization itself:Ÿ N-methyl-D-aspartate (NMDA) receptor blockersŸ Calcium channel alpha(2) ligandsAcetaminophenAcetaminiphen acts on the periaquaductal gray, and thence serotonergic and noradrenergic neurons to increase descending inhibition.[13,32,43,44,46,47]Serotonin- and Norepinephrine-Reuptake InhibitorsSerotonin re-uptake inhibitors (SSRI) and the serotonin precursor tryptophan enhance descending inhibition and are of use in stress-induced hyperalgesia. Serotonin and norepinephrine inhibitors (SNRI) and tricyclic anti-depressants (TCA) have a dual action, affecting both spinal activity and descending inhibition via monoamine pathways in the brain. The overall effect of these drugs is to enhance sleep and coping; reduce anxiety, and catastrophizing, all of which impact upon the neuromatix and ameliorate the pain experience.[5,13,42,43,48]In addition, TCAs are also thought to have a beneficial neuro-hormonal impact, and potentiate endogenous opioids. [42]Opioids and TramadolThe use of opioids for chronic pain is open to some controversy. From one perspective, opioids have the capacity to target critical mechanisms of central sensitization. Unfortunately, they not only target pain mechanisms, but also have significant impact upon immune function (as does central sensitization), and contribute to interleukin-1 mediated inflammatory pain hypersensitivity.[43,49-51] Taken with the significant positive reinforcement of these drugs on the reward centers of the brain, use of opioid therapy for clinical pain remains a case-by-case judgment in carefully selected and well-monitored patients.[44,52]Tramadol, an opioid-like drug, is of interest because of its multi-modal action. Though its binding at mu receptor cites is low, it is also a reuptake inhibitor of both serotonin and norepinephrine.[42]N-methyl-D-Aspartate Receptor BlockersActivation of NMDA-receptors (and protein kinase C) will induce hyperalgesia, supraspinal facilitatory loops, and apoptosis in the dorsal horn. NMDA blockers (ketamine, memantine and dextromethorphan) have been applied to interrupt this process, and may be analgesic in some circumstances, however theyhave a fairly narrow therapeutic window.[53-55]Calcium Channel Alpha(2)Delta LigandsVoltage-sensitive Calcium channels at the junction of primary afferent and second order sensory neurons will sustain an enhanced release of neurotransmitters in chronic pain. Gabapentin and pregabalin interrupt this action, and reduce the release of glutamate, norepinephrine and substance P, leading to reduction of both pre-, and postsynaptic noxious transmission. This, in turn, reduces the noxious input to the sensitized medulla. It would follow that in those cases of stress induced central sensitization, without a nociceptive component, calcium channel ligands may be less efficacious.Clinically it is common for these patients to cease responding to this class of drugs. Interestingly, there is some evidence that nociceptive inflammatory pain that is resistant to gabapentin, may respond to its second-generation analogue, pregabalin. Though working through somewhat different pathways some clinicians have recommended the use of carbamazepine, and its second-generation analogue oxcarbazepine, in these resistant cases. Evidence is limited for these substitutions.[42,47,54]
3.1.2. Non-pharmacological Approaches
- Each influence upon the neuromatrix offers an opportunity to target therapeutic interventions. While some of these targets are best addressed pharmacologically, a large number of opportunities are available for non-pharmacological therapies (NPT). For clinical purposes these NPT, like the pharmacological interventions, can be divided into two broad operative groups: Reducing CS itself; or responding to the effects of CS. In practice some of these interventions may well do both, though to varying degree.Repetitive Transcranial Magnetic Stimulation (rTMS)This relatively safe and non-invasive technique involves stimulation of the motor cortex and prefrontal cortex. It has seen limited application due to the short duration of its effects and significant equipment costs. It is interesting to note its greater efficacy in centrally, rather than peripherally, originated pain. In theory it reverses intra-cortical motor dysfunction, altering sensory-discriminative function, leading to restoration of descending inhibition, and also improvement of cognitive function.[56,57] However, some investigators argue that the analgesic effects are independent of descending inhibitory control and are influenced by other elements of the neuromatrix.[58] Percutaneous Electroneural Stimulation (PENS) Direct access to peripheral branches of multiple cranial and cervical nerves is available via percutaneous placement of electrodes on the human auricle. Utilizing a small, disposable, integrated circuit controlled, power source the patient can discreetly wear these devices for extended periods. The resulting stimulation of, primarily, the trigeminal, vagus and occipital afferents produce a number of changes in the neuromatrix. Among the observed results of PENS therapy are improved autonomic regulation, significant improvement in centrally mediated pain, sensory - discriminatory functions, serotonin/norepinephrine production and utilization, and endorphin production. The resultant analgesia and mood improvement appears to follow a ‘learning curve’[59], with patient improvement being graphically additive and cumulative based upon total wearing time of the device. Most of the observed improvement is putatively ascribed to changes in the function of the trigeminal complex and anterior cingulate portions of the neuromatrix; two of the major participants of CS. As such, PENS is one of our few cost-effective, non-invasive, low co-morbidity options for the treatment of central sensitization itself, as opposed to responding to the deleterious effects of CS.[1,2,5, 6,8-10,12-15,20-23,25-28,42-44,53, 55]The other broad category of non-pharmaceutical CS intervention encompasses manual therapy, virtual reality, improving stress tolerance, and transcutaneous electric nerve stimulation. These can be thought of as addressing the functional deficits created by the CS. In addition, some complementary and alternative approaches are being investigated and show promise, but evidence regarding their safety and efficacy is limited.Though primarily addressing the aftermath of CS, any therapy that reduces the overall burden on the neuromatrix will reduce the self-sustaining feedback that contributes to the progressive nature of CS. Manual TherapyManual therapy improves function and additionally produces an improvement in descending inhibition, which results in widespread analgesia. However, the analgesia is of short duration and provides limited assistance in desensitizing the neuromatrix. As such, manual therapy, from a clinical perspective, is an intervention to address functional rehabilitation.[4,13,60-64] Virtual RealityThough there is limited evidence, virtual reality has gained some support. Its effect is thought to derive from distraction in the hyper-vigilant patient, and would thus have potential benefit in patients with movement associated nociceptive etiology. It is not in widespread use and its clinical application is currently limited.[13,65]Improving Stress Tolerance and Neuro feedback Training Stress is a major etiologic and exacerbating factor for CS. Both endogenous (chronic pain) and exogenous (psychosocial changes) sources of stress can be involved. This often leads to clinical observation of the irritable, hyper-excitable chronic pain patient and is related to sympatho-afferent coupling in the hypothalamic-pituitary-adrenal axis of the neuromatrix. Additionally there are neuro-immune changes from upregulated pronociceptive immune mediators in primary afferent nociceptors. Reduction of stress levels can significantly improve the patient’s pain threshold, their maladaptive behavioral responses and autonomic balance. [12,14,17,35,36,66-68]Transcutaneous Electrical Nerve StimulationTENS is commonly employed in chronic pain states. Its effectiveness is related to activation of polysegmental inhibitory feedback, and additional elements of CS. It has some significant effect with focal, segmental chronic pain, however the results in widespread pain are equivocal.[13,40, 69-73]
3.2. Prognosis
- CS is a potentially disabling and devastating disease whose prognosis is dependent upon the ability to identify and address the initiating and perpetuating pathways. The possibilities here fit into three classes: Those derived from transformed nociceptive pain; those derived from stress; and most commonly, those representing various combinations of the two.In the case of transformed nociceptive pain, three elements are necessary for development of chronic clinical pain: neuroplasticity, peripheral sensitization, and central sensitization.[1] Neuroplasticity and peripheral sensitivity enable the central sensitivity. Removal, or prevention, of either or both of these, will improve the prognosis. This is best accomplished with adequate anesthesia, analgesia and inflammation control in the acute phase; early psychosocial intervention; and thorough patient education.[2,61,62,74]Unfortunately, once initiated central sensitization will engender additional presentations (Figure 1.), which increase the frequency and intensity of pain, and thereby increase the endogenous stress levels. These elevated stress levels will eventually result in increased sympatho-afferent coupling, and autonomic dysfunction; leading to anxiety, poor sleep, difficulty coping, and lowered pain thresholds; and perhaps more importantly, an increased risk of developing additional presentations of CS.[2,74]Once the CS reaches this stage, mere removal of the initiating stimulus will not insure favorable outcomes. A corollary to this: Patients with persistent biological sources of nociception, certain inflammatory diseases for instance, will have a continuing stimulus for the development and/or maintenance of CS and are likely to experience an extended disease course, including the development of additional CS presentations (syndromes), and the focus devolves to dealing with the effects of the CS, rather than control or eradication of the CS.[2,3,74]In the case of stress induced CS, no biological axis may exist in the early stages. However, maladaptive behavior may engender biological issues that will contribute to maintenance and exacerbation of CS. The chronic pain associated with PTSD and depression in the apparently healthy individual may well be examples of just this sequence.
4. Conclusions
- Chronic clinical pain associated with CS, is a potentially progressive, devastating, multimodal disease with a significant worldwide economic and social burden. Effective intervention is dependent upon recognizing the fundamental differences in acute and chronic pain, the effects on and by the neuromatrix upon the biopsychosocial health of the individual patient, and integrating that knowledge into a comprehensive multidisciplinary therapeutic plan. Its prognosis is guarded.
ACKNOWLEDGEMENTS
- This paper was prepared in partial fulfilment of requirements for the MSc degree in pain management at University of Edinburgh, College of Medicine and Veterinary Medicine, Edinburgh, Scotland.