-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2026; 16(1): 1-20
doi:10.5923/j.chemistry.20261601.01
Received: Nov. 30, 2025; Accepted: Dec. 20, 2025; Published: Jan. 7, 2026

DFT Analysis of the Gas Phase Interactions of Choline Chloride with Urea
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLDan E. Diendere1, Edouard Tapsoba2, Françoise Diendere1, Moussa R. Bougouma1
1Laboratoire de Chimie Analytique et de Physique Spatiale (LAC@PSE), Université Norbert ZONGO, Koudougou, Burkina Faso
2Laboratoire de Chimie Analytique, Environnementale et Bio-Organique (LCAEBIO), Université Joseph KI-ZERBO, Ouagadougou, Burkina Faso
Correspondence to: Françoise Diendere, Laboratoire de Chimie Analytique et de Physique Spatiale (LAC@PSE), Université Norbert ZONGO, Koudougou, Burkina Faso.
| Email: |  |
Copyright © 2026 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Choline chloride and urea interact to form a Deep Eutectic Solvent widely used as a green solvent in several applications such as electrochemistry, chemical substance separation and reactions. We searched for the equilibrium geometries, charges distributions of model compounds of their respective associated complexes of 1:1, 1:2 and 1:3 ratios. The calculated interaction energies indicate that several stable conformers of the DES may form and coexist in the solvent medium. Beside coulombic interactions, NBO analysis shows that the through bond interactions between occupied orbitals and empty antibonding molecular orbitals contribute to spread the electronic charge and stabilize the systems. These interactions involve mainly the lone pairs of the electronegative elements Cl, N and O so that H-bonds of the type C=O---H-O, Cl---H-N, Cl---H-C and C=O---H- link the various neutral and ionic fragments in the model compounds of the solvent. Calculated Wiberg bond indices revealed the partial covalent nature of these H-bonds.
Keywords: Density functional theory calculations, Choline chloride-urea compounds, Deep eutectic solvents, NBO analysis, H-bonds
Cite this paper: Dan E. Diendere, Edouard Tapsoba, Françoise Diendere, Moussa R. Bougouma, DFT Analysis of the Gas Phase Interactions of Choline Chloride with Urea, American Journal of Chemistry, Vol. 16 No. 1, 2026, pp. 1-20. doi: 10.5923/j.chemistry.20261601.01.
Article Outline
1. Introduction
- The investigations on safe solvents that protect ecosystems nowadays have paved the way to the synthesis in 2001 by Abbott and coworkers [1-2] of novel mediums called deep eutectic solvents (DES) or low temperature transition mixtures (LTTM). Since then, these solvents have gained great interest and continuing research is done on their development and the understanding of the physical and chemical properties that they display. They are liquid over a certain range of temperatures, have negligible volatility as compared to traditional solvents such as organic solvents and show good thermal stability and electrical conductivity. As such, they are candidates for reaction mediums in the fields of material science, electrochemistry and biochemistry [3-7]. These important fields of application of DESs require that their geometric structures and their electronic charge distributions as well as their physico-chemical properties be well defined in order to offer optimal conditions for their use.A deep eutectic solvent (DES) is defined as a mixture of two or more components which has a temperature of its eutectic point significantly below that of its ideal mixture. Studies done on the solid-liquid phase diagrams have shown that such mixtures present negative deviation from ideality at their eutectic point with specific ratio of the components and are liquid at operating temperatures over a certain range of composition [2,8-9]. They are mainly mixtures of Lewis or Bronsted acids and bases, which may be neutral or ionic. DESs that do not contain metal ions or metal free DES [10] are well established and an example of this category of solvents is the mixture of choline chloride and urea in a 1:2 ratio. This solvent contains a quaternary ammonium salt and urea as interacting agents. It is argued that the lowering of the eutectic point below the ideal one is due to interaction between the species through H-bonding so that the preparation of these solvents implies appropriate selection of the H-bond donor and acceptor. For the particular case of the metal-free DES made of choline chloride (ChCl) and urea (U), several experimental and theoretical studies have been done in attempts to characterize it and to understand its properties. Abbot and al. [2] have studied the phase diagram constructed from measurements of freezing points and showed that the eutectic point of the solution has a temperature of 12°C with a composition of 1:2 ratio of respectively choline chloride and urea. At this composition, the mixture is referred to as reline. By using DSC technique, Morrisson et al. [11] have obtained an eutectic point of 17°C and a similar composition of the mixture. Later on, Meng and coworkers [12] have done a more extensive study on the phase diagram of this DES by measuring the melting point of different mixtures containing known amounts of water via DSC and optical microscopy techniques. Their results indicate that the presence of water greatly influences the interactions between the two components and therefore the physico-chemical properties of the DES. At the eutectic composition ratio of Abbot and al., their measured melting temperature was 25°C. The difference in the various measured eutectic temperatures is explained by the presence of water in the DES, as the constituents are hygroscopic. Water induces formation of new interactions with the polar constituents and therefore affects the density, viscosity and electrical conductivity of the mixture. These authors suggested that properties of DESs be measured on water free samples. The formation of H-bond in water free mixtures has been suggested from the liquid phase neutron diffraction measurements of isotopically substituted samples by Hammond et al. [13] and the IR and NMR investigations of Perkins et al. [14-15]. Ashworth et al. [16] have done a theoretical study of pairwise interactions between the constituents and of clusters of Cl- with urea. This extensive work on conformers of pairs of species by DFT has revealed a network of H-bonding within the pairs of neutral and ionic species. They calculated interaction energies of these conformers in order to understand the basis of the H-bond formation. In this interesting analysis of the interactions of pairs of fragments of the DES, geometries and charge distributions have not been detailed to link the structures to the observed H-bonding. The present work is a continuation of the study on the interactions between choline chloride and urea based on model systems composed of several ratios of these constituents. We report calculated geometrical structures of urea, choline chloride and their associated compounds. We search for the gas phase optimal geometries and charge distributions of the constituents and the model compounds of reline that would enlighten our understanding of their structural, bonding and physico-chemical properties.
2. Computational Methodology
- The work consists of a DFT [17] study of the geometries, energies and electronic structures of the fragments choline chloride and urea as well as those of their successive associations to form model compounds of reline. The GaussView06 software [18] was used to construct and visualize the various systems. Then optimized geometrical parameters, energies and charge distributions were carried out by the Gaussian 09 software package [19] by using the gradient corrected hybrid functional BYL3P [17] along with the standard 6-31+G(d,p) basis set which is a split valence band basis with polarization and diffuse functions to well describe bond formation and charge delocalization. No symmetry constraints were maintained during the optimization of the compounds. Vibrational frequencies were calculated at each optimized structure to characterize it as a minimum. The total energies are all ZPE corrected. The mechanism of charge delocalization through bonds in these systems was studied by performing NBO calculations on the optimized structures with the NBO codes provided by the Gaussian 09 package. Atomic charges were also calculated by using the NBO subroutine. The covalent nature of the bonds was analyzed by calculating Wiberg bond indices in the optimized species.
3. Results and Discussion
3.1. Geometries and Energies
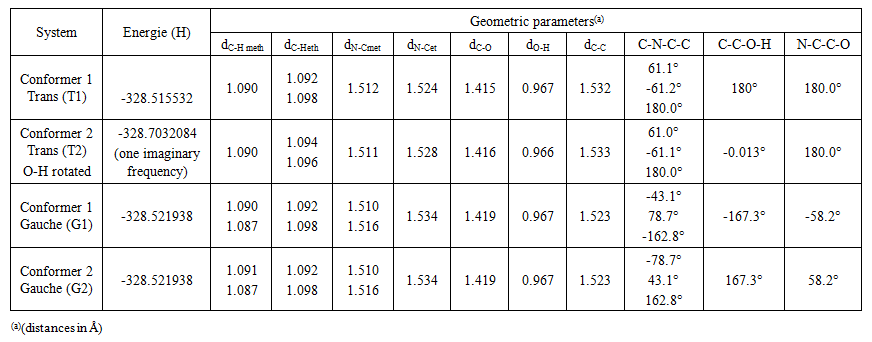
- The model compounds of the DES reline were investigated by starting with a search of the minimum structures of the fragments urea and choline chloride. Table 1-3 contain respectively the energies and optimized geometrical parameters of urea CO(NH2)2 (or U), its dimers, the choline cation [(CH3)3N(CH2)2OH]+ (or Ch+), choline chloride [(CH3)3N(CH2)2OH]Cl (or ChCl) and experimental results on urea. Figure 1-3 display their respective optimized structures. a. Urea monomers and dimers The equilibrium structure of urea is basis set dependent and our results in Table 1 and Figure 1a-c indicate that the conformation of C2 symmetry is its stationary structure while the others of C2V and Cs symmetries are respectively second and first order saddle points. The dihedral angles OCN3H4, OCN3H5 and N6CN3H4 in the C2 minimal energy conformer have respective values of 11.8°, 152.3° and -168.2°. These parameters along with the calculated bond lengths and bond angles reproduce quite well the microwave results of Godfrey and al. [20] as well as the gas phase data obtained at the MP2/6-31G(d, p) and MP2/6-311G(d,p) by Raptis and coworkers [21]. This attests of the good quality of the basis set used in our approach. Oher experimental geometries of urea from neutron diffraction and X-rays analysis gave a planar structure with C-O and C-N bond lengths of 1.265Å and 1.349 Å respectively [22-24]. This CO bond length obtained in solid state is longer than our calculated gas phase value and may be due to intermolecular interactions. The structures of dimers of urea have been also explored and are shown on figure 1.d-f. They are true minima and the interaction energies, which range from -50.6 to -37.8 kJ.mol-1 indicate that they are stable gas phase structures. One may notice that the interactions between two urea molecules cause elongation of the C-O and N-H bonds and opening of the C-N-H angles of interacting groups. The structural arrangements of these dimers in figure 1d-f well illustrate the proton donor and acceptor ability of urea through its N-H bonds and the lone pairs of electrons on its N and O atoms. One therefore expects urea to play a central role in the formation of compounds with choline chloride.
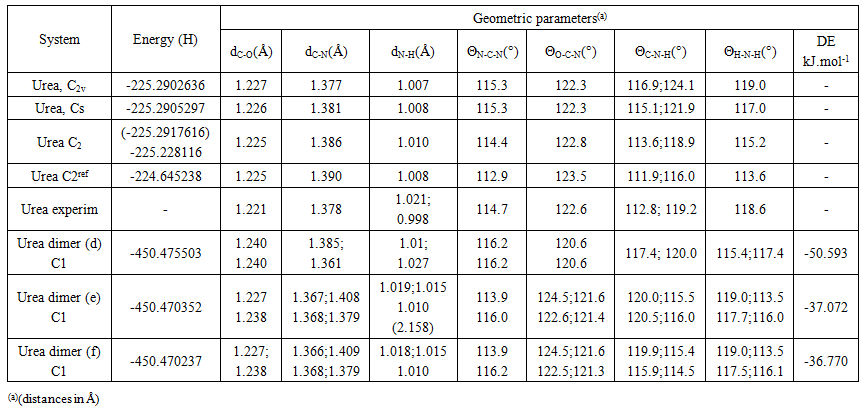 | Table 1. Energies and geometries of optimized urea monomers and dimers |
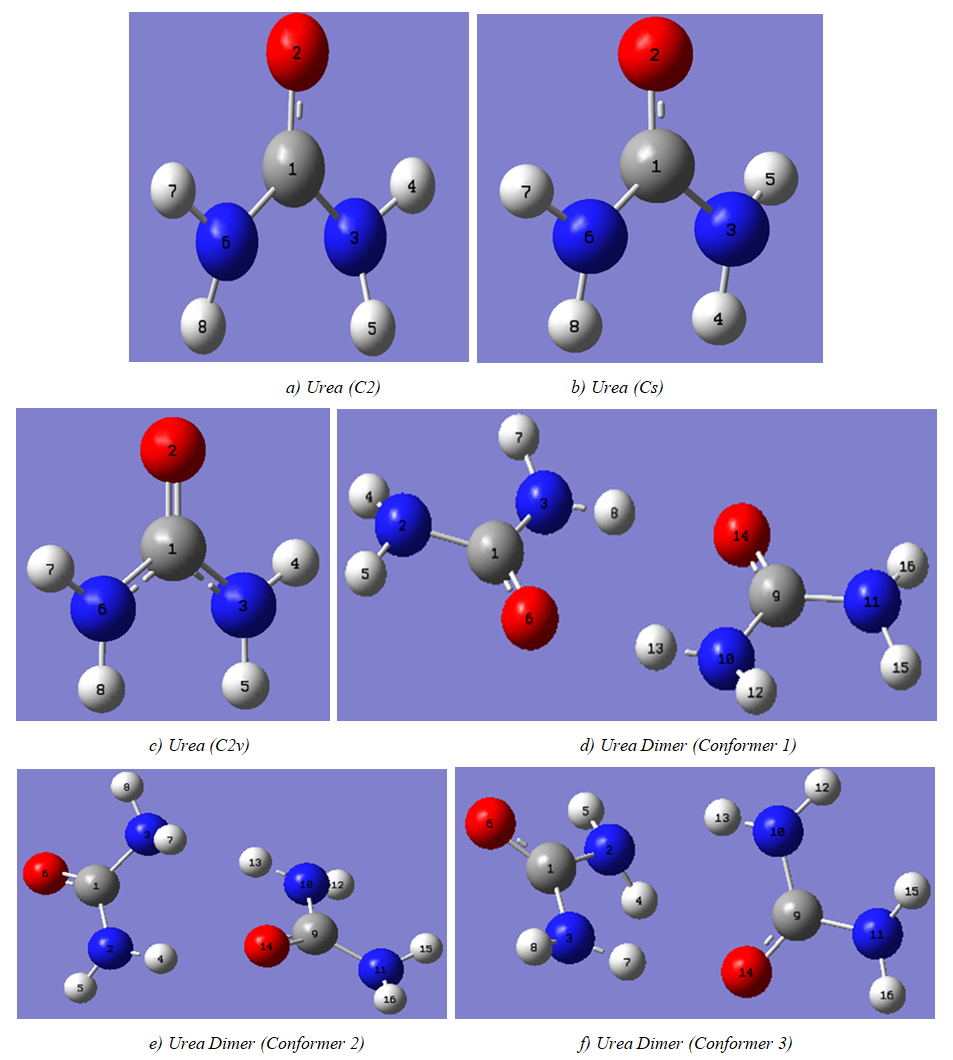 | Figure 1. Optimized structures of urea monomers and dimers |
 | Table 2. Energies and geometries of optimized choline cation monomers |
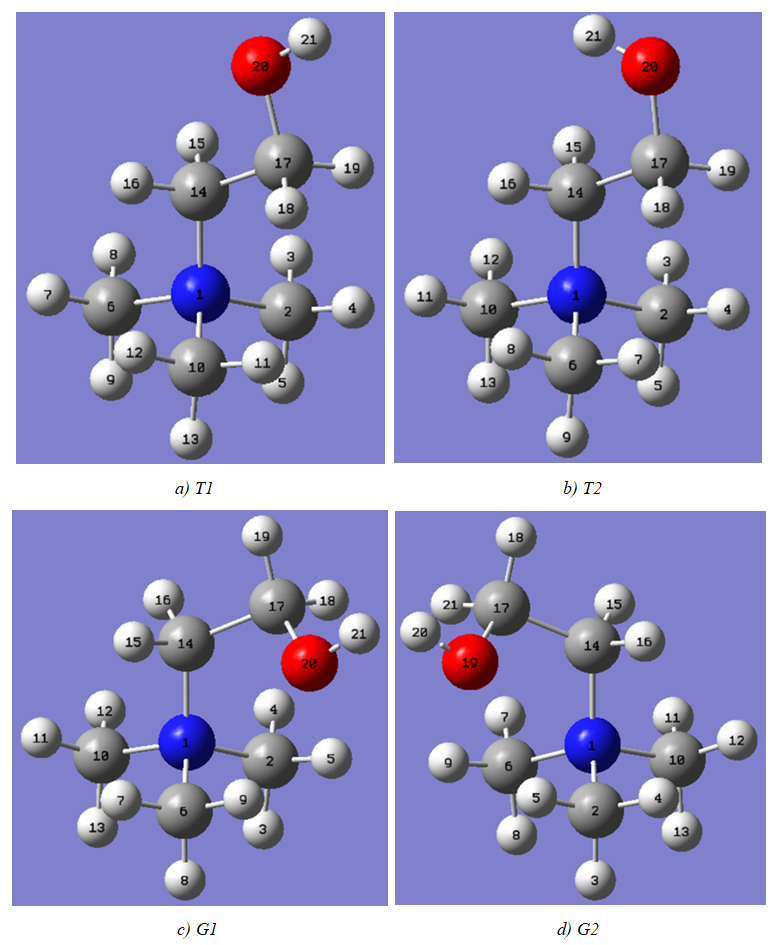 | Figure 2. Optimized structures of Conformers of the cholinium cation |
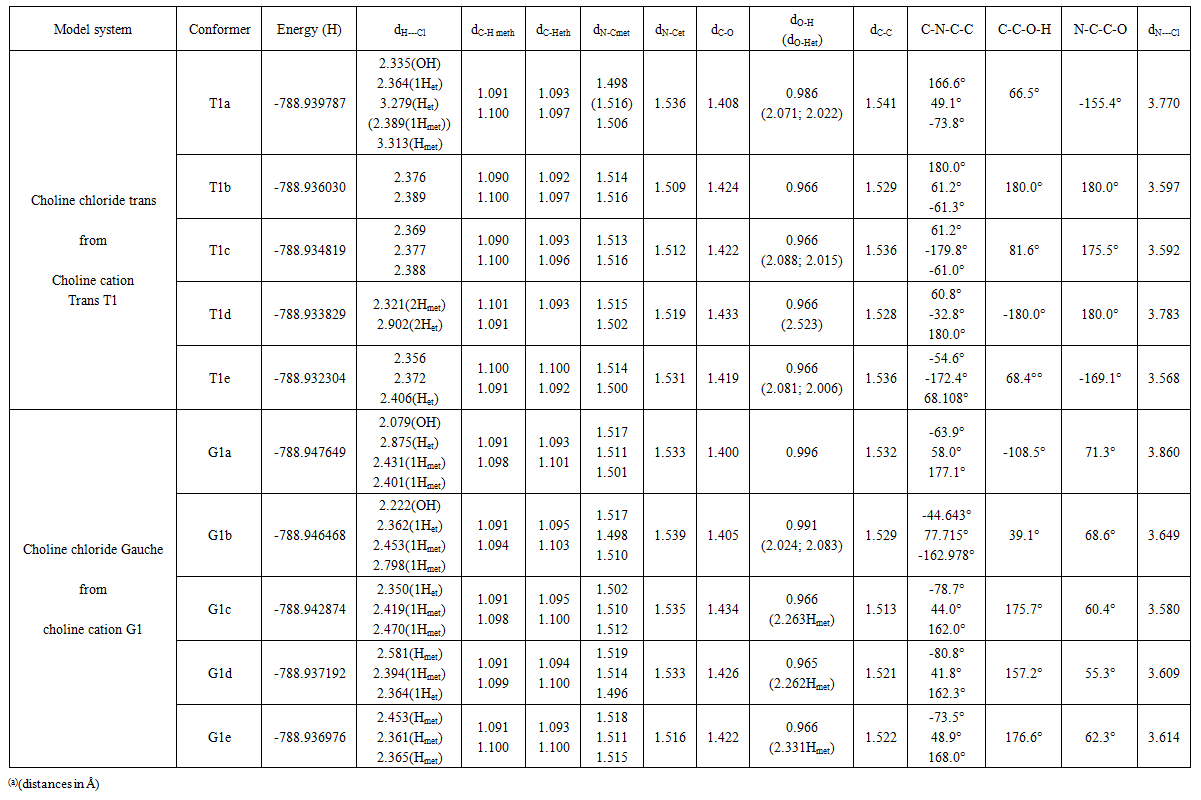 | Table 3. Energies and geometries of optimized choline chloride monomers (a) |
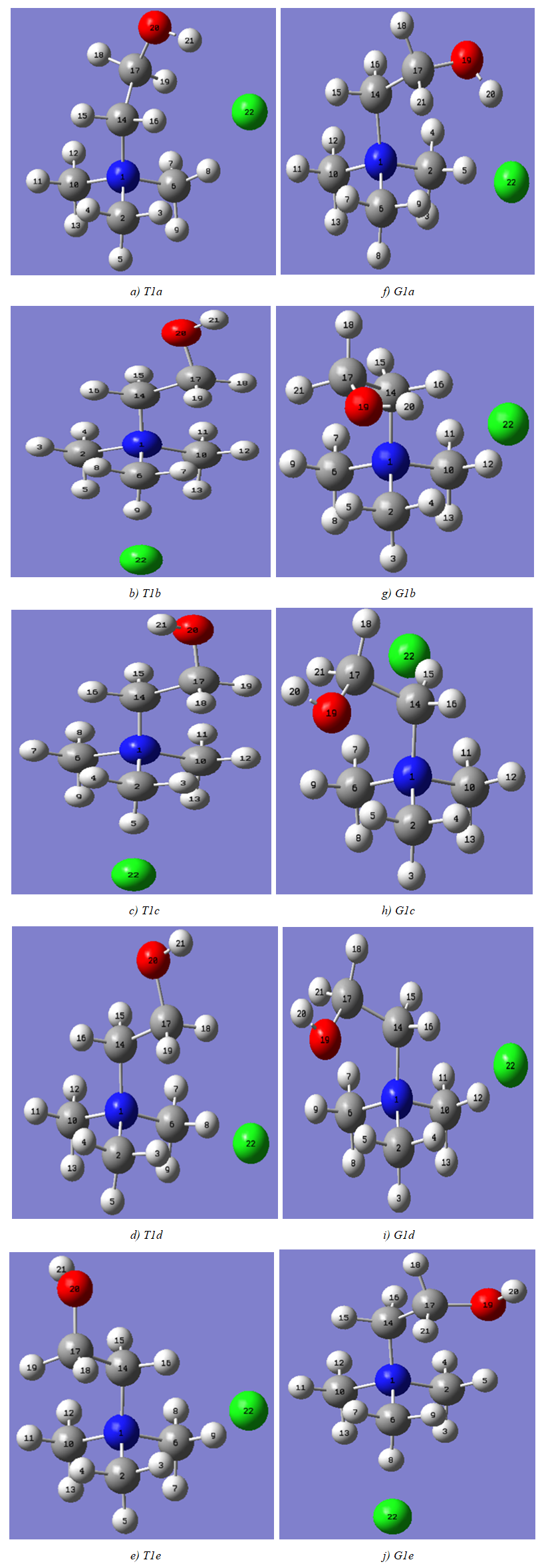 | Figure 3. Optimized structures of conformers of choline chloride |
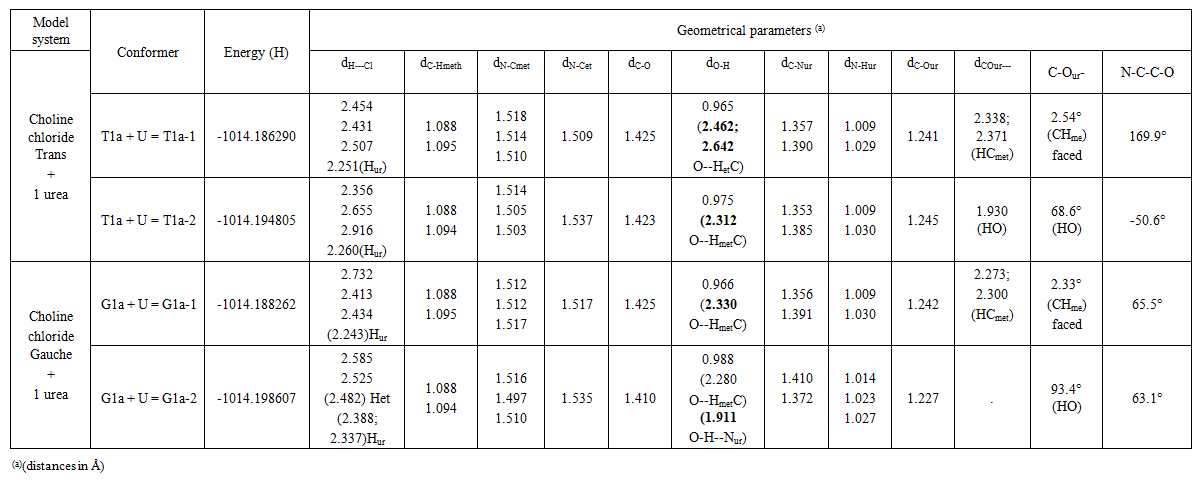 | Table 4. Energies and geometries of optimized systems of choline chloride and 1 urea |
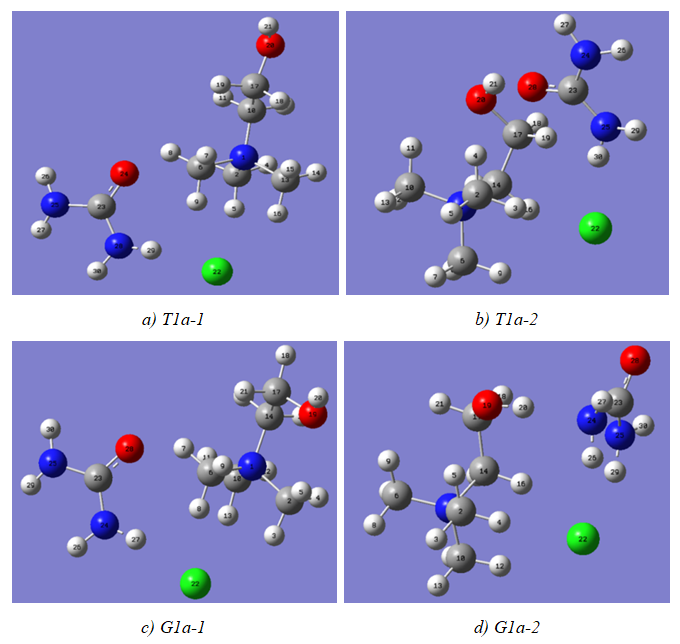 | Figure 4. Optimized conformers of 1:1 complexes of choline chloride and urea |
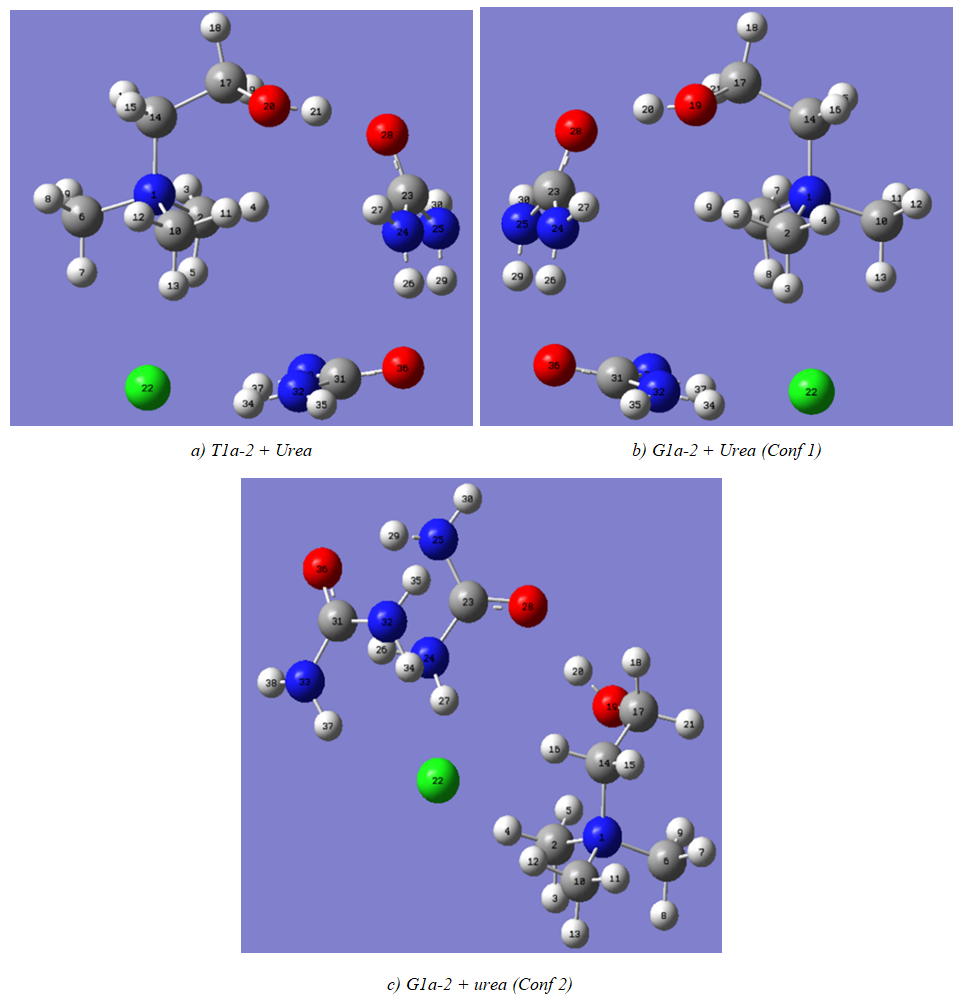 | Figure 5. Choline chloride complexes with 2 ureas |
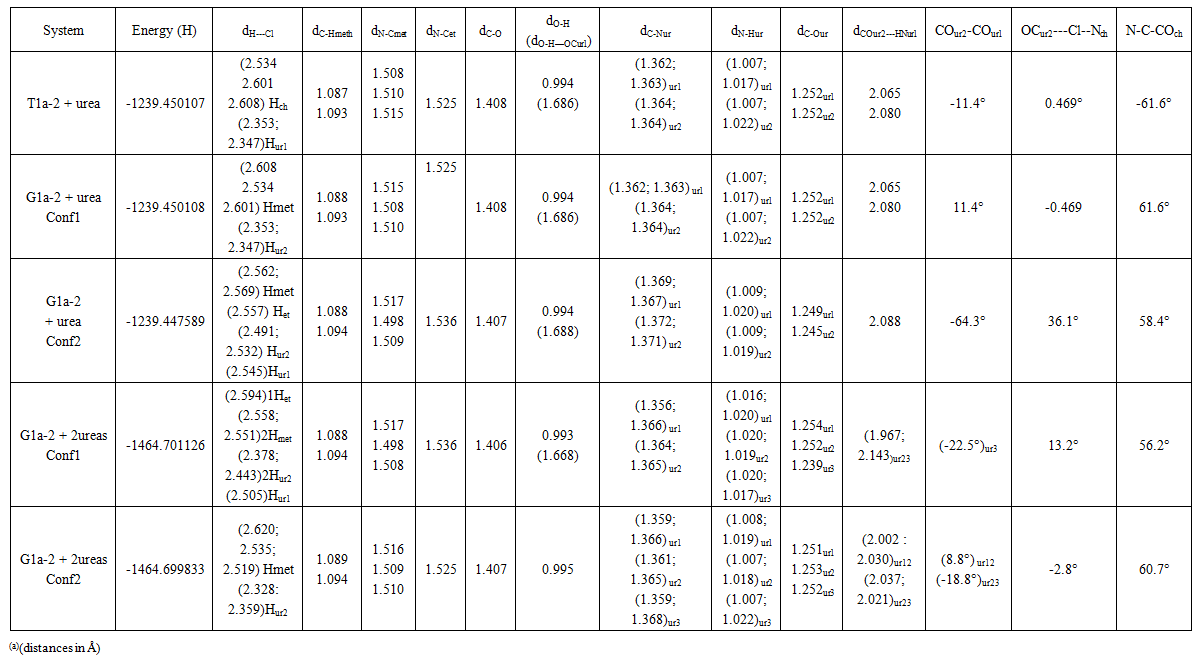 | Table 5. Energies and geometries of optimized systems of choline chloride with 2 or 3 ureas (a) |
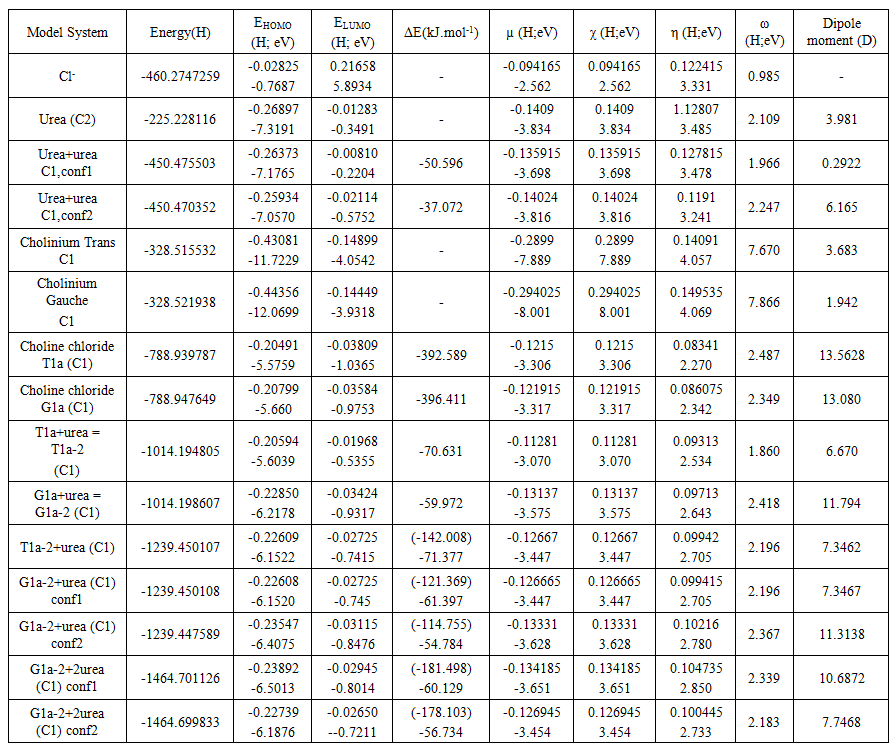 | Table 6. Reactions energies and quantum chemical parameters |
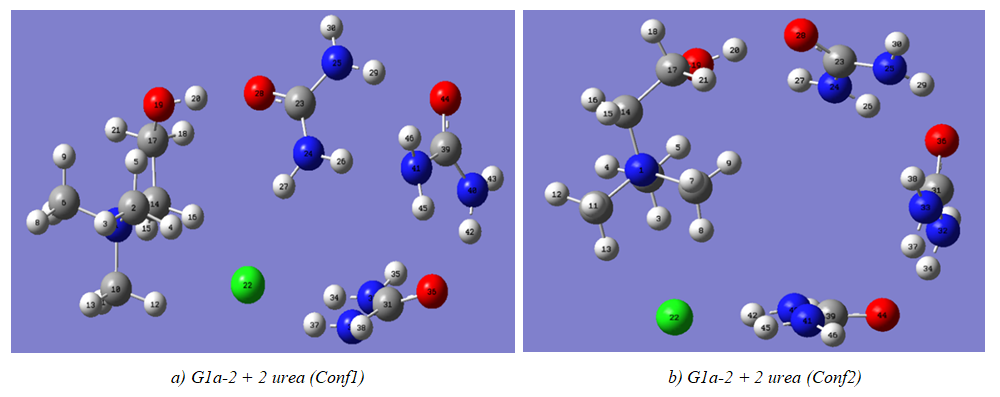 | Figure 6. Choline chloride complexes with 3 ureas |
3.2. Charge Distributions
- a. Atomic chargeIn the natural atomic orbital (NAO) basis, the natural population on an atom (A) is the sum over all contributing NAOs of the natural population qi(A) of orbital φi(A). The difference between its atomic number and its natural population is its atomic charge Q(A). According to the Pauli principle, 0 ≤ qi(A) ≤ 2. Table 7 displays the calculated atomic charges given by the present analysis. One may note the negative charge carried by the carbon atoms of the choline cation and which increases upon addition of Cl- and urea due to the interactions. The results also indicate the charges on the electronegative elements N and O, which follow the same trend as the carbon atoms. Concurrently, there is a decrease in charge on the chlorine atom from -1.0 a.u to -0.856 au in the most stable 1:2 ratio model system. The H atoms are all polarized and those of the methyl groups nearest to the chloride ion have up to +0.305 au in the most stable model of reline.
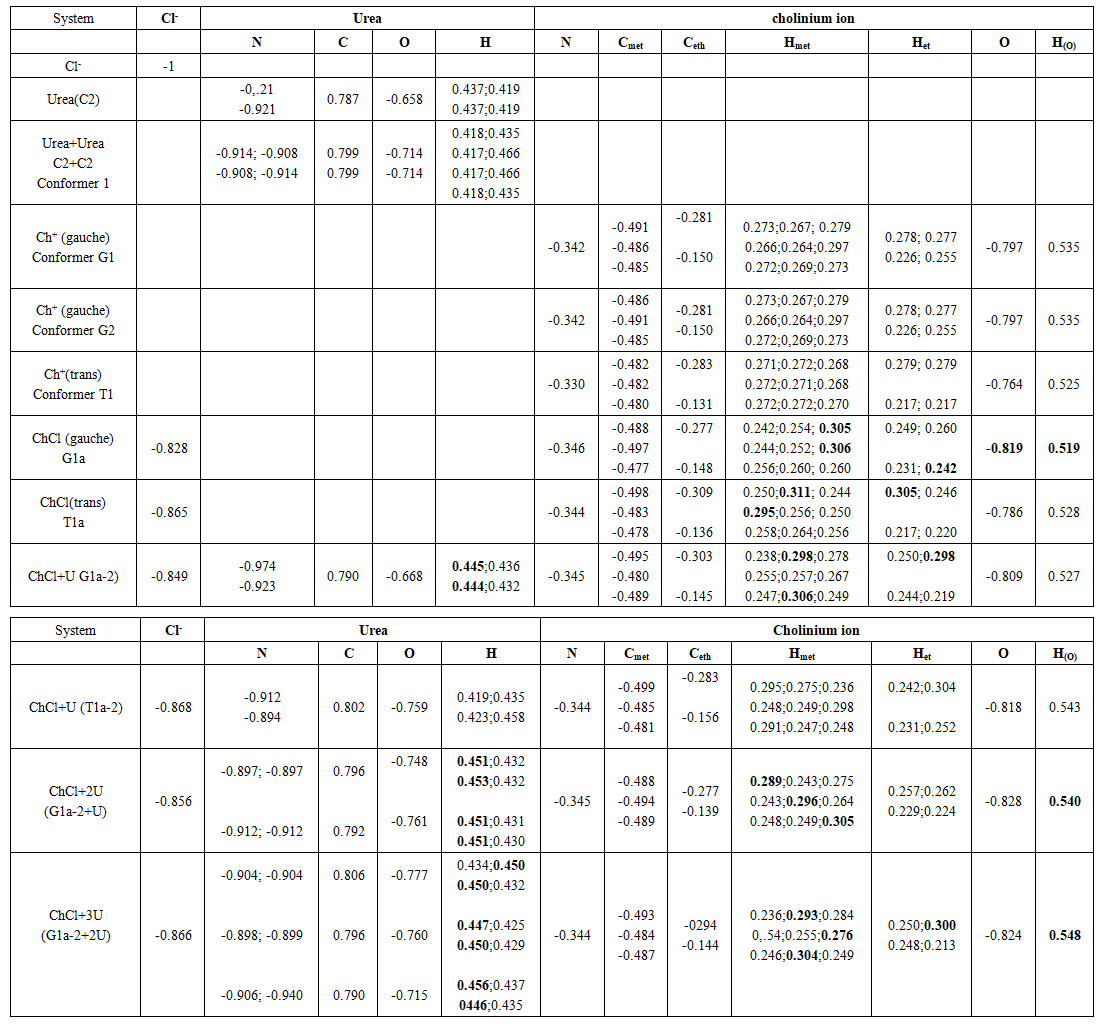 | Table 7. Atomic charges (in u.a) of the most stable compounds |
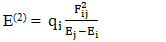 | (1) |
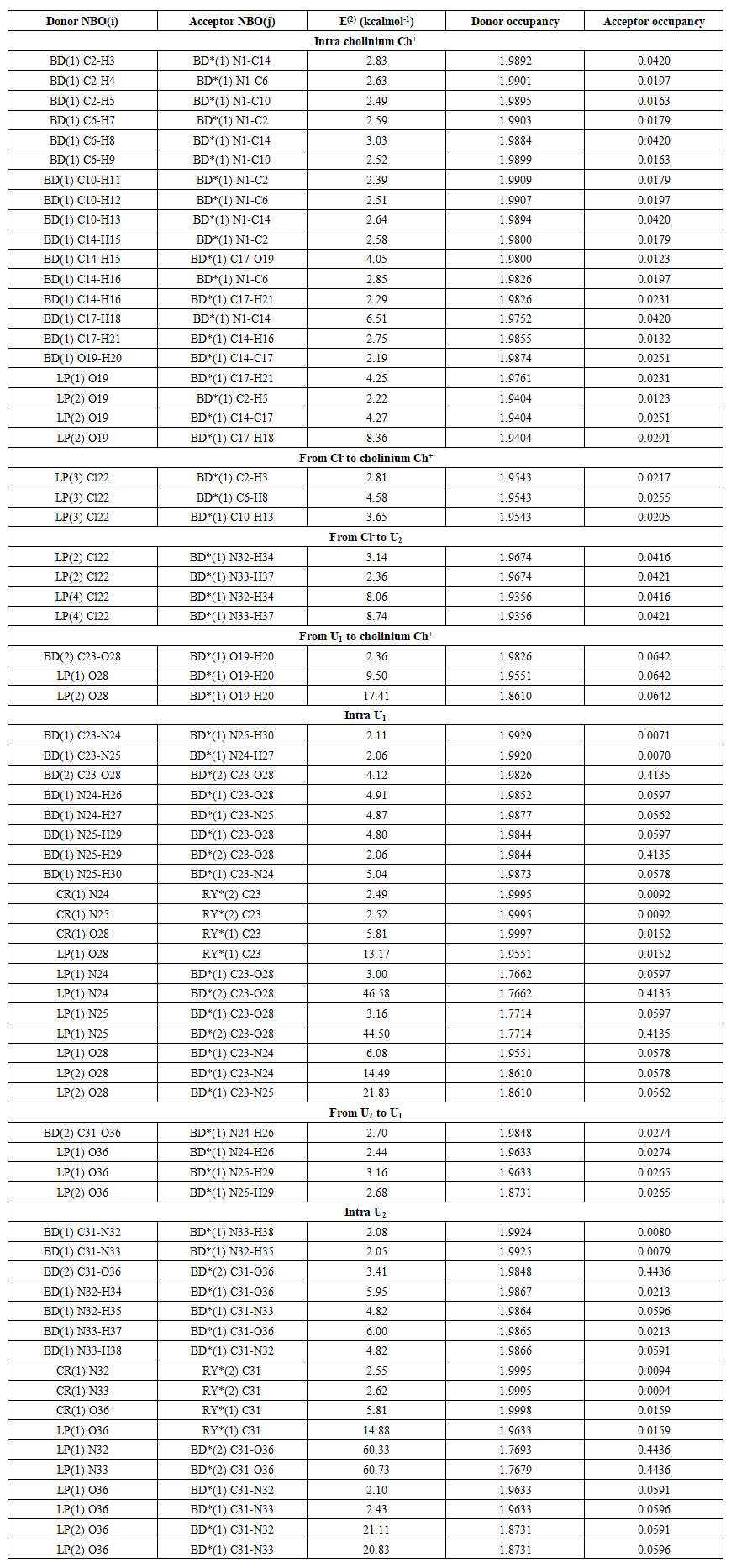 | Table 8. Second order perturbation energy E(2) in kcal.mol-1 for the most important charge transfer interactions in the most stable model of choline chloride plus two urea (E(2) ≥ 2 kcal.mol-1) |
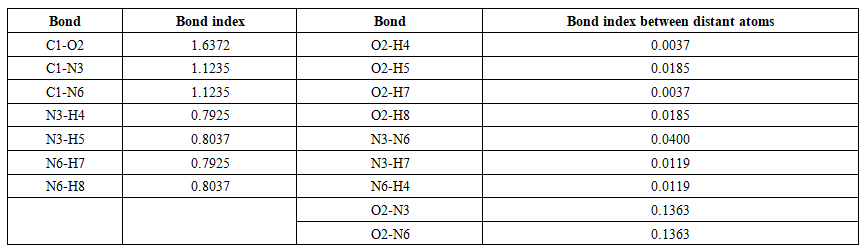 | Table 9. Wiberg bond Indices in the urea molecule (C2) |
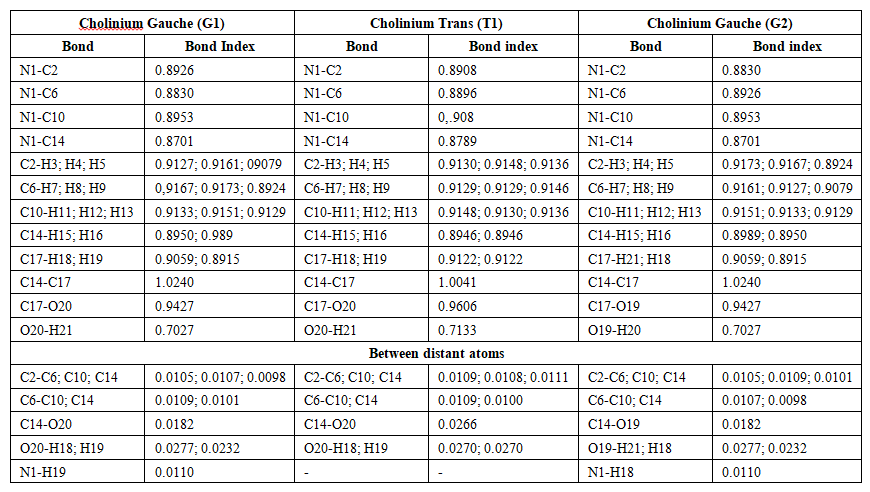 | Table 10. Wiberg bond indices in the cholinium cation Gauche and Trans (C1) |
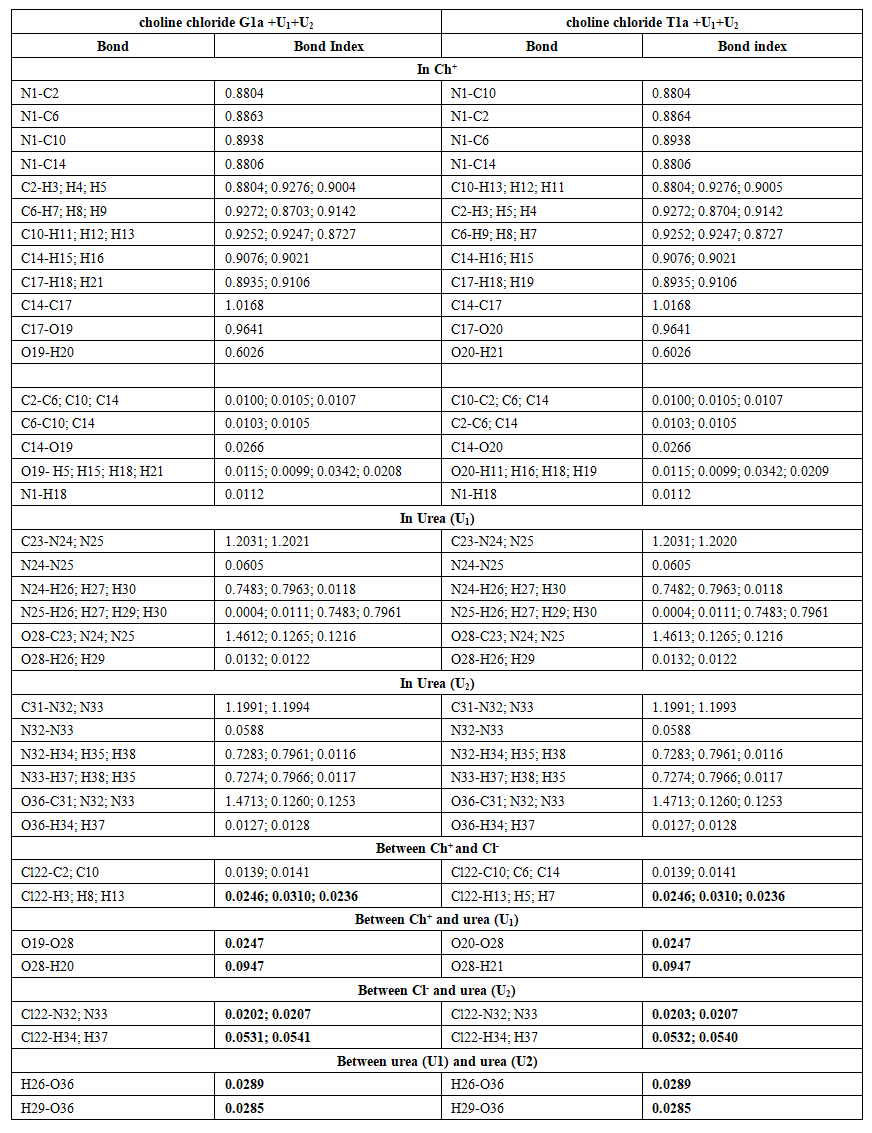 | Table 11. Wiberg bond Indices in choline chloride G1a et T1a plus 2 urea molecules (C1) |
3.3. Quantum Chemical Parameters of the Model Compounds
- The electronic chemical potential µ, the electronegativity χ, the chemical hardness η and the global electrophilicity index ω of the model compounds were determined in our study from the energies of the frontier orbitals EHOMO and ELUMO using the following expressions as suggested by other authors [28-30].
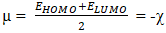 | (2) |
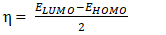 | (3) |
 | (4) |
4. Conclusions
- We report Density Functional Theory calculations for urea, Cl-, the choline cation, choline chloride and model compounds of the DES reline that may result from their association. The total ZPE corrected energies; detailed structural parameters, interaction energies, charge distribution and some quantum chemical parameters of all these systems are presented. Our results predict minimum energy structures for the DES in which partial covalent H-bonds link the ions and the molecules of urea in accord with previous work. Our study reveals the presence in the model solvent of H-bonds of the type C=O---H-O, Cl---H-N, Cl---H-C and C=O---H-N that result from the interactions of the fragments. Formation of these H-bonds maximizes the intra and intermolecular interactions and stabilize the system. Our data do not show evidence of a specific speciation of Cl- with urea although such geometries may minimize coulombic interactions. Our calculated quantum chemical parameters and the geometrical arrangement of the fragments in the model compounds also predict the 1:2 ratio in which choline chloride and urea combine in the DES.
ACKNOWLEDGEMENTS
- The authors are grateful to Pr Jean-Marc Sotiropoulos at the CNRS of Pau in France for his contribution to this work.