| [1] | J.-F. Le Page, Catalyse de contact: conception, préparation et mise en øeuvre des catalyseurs industriels. Editions Technip, 1978. |
| [2] | G. S. Kirchoff, «History of glucose syrups», Memoires L’Academie Imp. Sci. St Petersburg, vol. 4, p. 27, 1811. |
| [3] | B. H. Davis et W. P. Hettinger, Éd., Heterogeneous Catalysis: Selected American Histories, vol. 222. in ACS Symposium Series, vol. 222. Washington, D.C.: American Chemical Society, 1983. doi: 10.1021/bk-1983-0222. |
| [4] | L. Lloyd, Handbook of industrial catalysts. Springer Science & Business Media, 2011. |
| [5] | S. Cheballah et M. S. Dahmani, «Etude comparative des systèmes: Hétéropolyanionique et pérovskite dans la réaction de reformage du méthane par le dioxyde de carbone», PhD Thesis, UMMTO, 2018. |
| [6] | L. P. Blanchard et Y. Yim, «L’effet du molybdène sur l’oxydation partielle du méthane par l’oxygène-pur», Can. J. Chem. Eng., vol. 49, n° 4, p. 488‑494, août 1971, doi: 10.1002/cjce.5450490410. |
| [7] | O. Colin, «Plateforme pyridylalkylamine modulable: un outil pour la catalyse», PhD Thesis, Versailles-St Quentin en Yvelines, 2015. |
| [8] | J. A. Busby, «The activation and deactivation of platinum/rhodium catalysts for ammonia oxidation», 1976. |
| [9] | G. A. Somorjai, «The Catalytic Hydrogenation of Carbon Monoxide. The Formation of C 1 Hydrocarbons», Catal. Rev., vol. 23, n° 1‑2, p. 189‑202, janv. 1981, doi: 10.1080/03602458108068075. |
| [10] | O. B. J. Fraser, «Nickel as a Catalyst», Trans. Electrochem. Soc., vol. 71, n° 1, p. 425, 1937. |
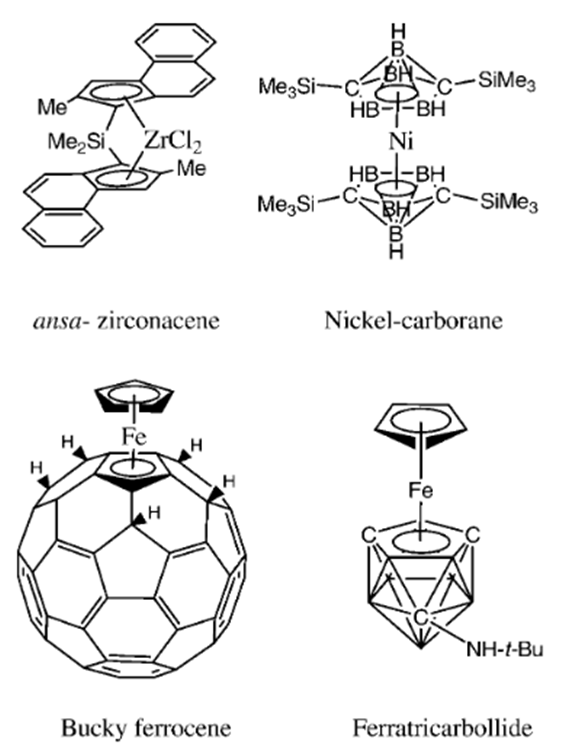
| [11] | T. J. Colacot et N. S. Hosmane, «Organometallic Sandwich Compounds in Homogeneous Catalysis: An Overview», Z. Für Anorg. Allg. Chem., vol. 631, n° 13‑14, p. 2659‑2668, oct. 2005, doi: 10.1002/zaac.200500224. |
| [12] | D. Astruc, «Chapitre 20 - Catalyse hétérogène», in Chapitre 20 - Catalyse hétérogène, EDP Sciences, 2021, p. 471‑508. doi: 10.1051/978-2-7598-1106-9.c025. |
| [13] | K. A. Ahrendt, C. J. Borths, et D. W. MacMillan, «New strategies for organic catalysis: the first highly enantioselective organocatalytic Diels- Alder reaction», J. Am. Chem. Soc., vol. 122, n° 17, p. 4243‑4244, 2000. |
| [14] | E. R. Jarvo et S. J. Miller, «Amino acids and peptides as asymmetric organocatalysts», Tetrahedron, vol. 58, n° 13, p. 2481‑2495, 2002. |
| [15] | O. Riant et H. B. Kagan, «Asymmetric Diels-Alder reaction catalyzed by chiral bases», Tetrahedron Lett., vol. 30, n° 52, p. 7403‑7406, 1989. |
| [16] | «Organocatalyse asymétrique: une solution de chimie de synthèse sélective et durable remporte le prix Nobel de Chimie 2021», CAS. Consulté le: 11 avril 2023. [En ligne]. Disponible sur: https://www.cas.org/fr/resources/blog/2021-chemistry-nobel. |
| [17] | K. Brand, «Chalcogen based organocatalysts in transesterification», PhD Thesis, 2015. |
| [18] | J. T. Richardson, Principles of catalyst development. Springer, 2013. |
| [19] | J. Lipkowski et P. N. Ross, «Electrocatalysis», 1998, Consulté le: 9 avril 2025. [En ligne]. Disponible sur: https://books.google.com/books?hl=fr&lr=&id=HZowkmoF4ngC&oi=fnd&pg=PR13&dq=electrocatalysis +review&ots=9c_qT661cl&sig=m-ghGaLy3phxNoeb9AbpE2HoHmo. |
| [20] | N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu, et H. M. Chen, «Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives», Chem. Soc. Rev., vol. 46, n° 2, p. 337‑365, 2017, doi: 10.1039/C6CS00328A. |
| [21] | C. Tanielian, «Decatungstate photocatalysis», Coord. Chem. Rev., vol. 178, p. 1165‑1181, 1998. |
| [22] | L. Marchetti et M. Levine, «Biomimetic Catalysis», ACS Catal., vol. 1, n° 9, p. 1090‑1118, sept. 2011, doi: 10.1021/cs200171u. |
| [23] | «Biomimetics - an overview | ScienceDirect Topics». Consulté le: 10 avril 2025. [En ligne]. Disponible sur: https://www.sciencedirect.com/topics/earth-and-planetary-sciences/biomimetics. |
| [24] | M. Bilal, M. Adeel, T. Rasheed, et H. M. N. Iqbal, « Multifunctional metal–organic frameworks-based biocatalytic platforms: recent developments and future prospects », J. Mater. Res. Technol., vol. 8, n° 2, p. 2359‑2371, avr. 2019, doi: 10.1016/j.jmrt.2018.12.001. |
| [25] | W. S. Koe, J. W. Lee, W. C. Chong, Y. L. Pang, et L. C. Sim, «An overview of photocatalytic degradation: photocatalysts, mechanisms, and development of photocatalytic membrane», Environ. Sci. Pollut. Res., vol. 27, n° 3, p. 2522‑2565, janv. 2020, doi: 10.1007/s11356-019-07193-5. |
| [26] | J. Lipkowski et P. N. Ross, Electrocatalysis. John Wiley & Sons, 1998. |
| [27] | G. Valenti et al., «Co-axial heterostructures integrating palladium/titanium dioxide with carbon nanotubes for efficient electrocatalytic hydrogen evolution», Nat. Commun., vol. 7, n° 1, p. 13549, 2016. |
| [28] | T. R. Ward et C. Copéret, «Introduction: Bridging the Gaps: Learning from Catalysis across Boundaries», Chem. Rev., vol. 123, n° 9, p. 5221‑5224, mai 2023, doi: 10.1021/acs.chemrev.3c00029. |
| [29] | «Catalyse homogène, hétérogène et enzymatique», MAXICOURS. Consulté le: 21 mars 2023. [En ligne]. Disponible sur: https://www.maxicours.com/se/cours/catalyse-homogene-heterogene-et-enzymatique/. |
| [30] | «Catalyseur en chimie: Cours et explications │ StudySmarter», StudySmarter FR. Consulté le: 22 février 2024. [En ligne]. Disponible sur: https://www.studysmarter.fr/resumes/physique-chimie/chimie/catalyseur/. |
| [31] | G. J. Hutchings, «Heterogeneous catalysts—discovery and design», J Mater Chem, vol. 19, n° 9, p. 1222‑1235, 2009, doi: 10.1039/B812300B. |
| [32] | P. McMorn et G. J. Hutchings, «Heterogeneous enantioselective catalysts: strategies for the immobilisation of homogeneous catalysts», Chem. Soc. Rev., vol. 33, n° 2, p. 108, 2004, doi: 10.1039/b200387m. |
| [33] | O. Roelen et A. G. Ruhrchemie, «A process for the preparation of oxygencontaining compounds», Pat., vol. 849548, 1938. |
| [34] | H. S. Eleuterio, «Olefin metathesis: chance favors those minds that are best prepared», J. Mol. Catal., vol. 65, n° 1‑2, p. 55‑61, 1991. |
| [35] | H.-B. Lee et P. M. Henry, «Oxidation of olefins by palladium(II). VIII. Kinetics of the oxidation of ethylene by palladium(II) chloride in methanol», Can. J. Chem., vol. 54, n° 11, p. 1726‑1738, juin 1976, doi: 10.1139/v76-246. |
| [36] | C. Thomas, «New multifunctional ligands for the catalytic carbonylation of methanol», 2002. |
| [37] | S. Jeulin, «Synphos et difluorphos: diphosphines chirales par atropoisomérie. Évaluation des propriétés stériques et électroniques, synthèse d’analogues et applications en catalyse asymétrique», PhD Thesis, Chimie ParisTech, 2005. |
| [38] | «Chapitre 19 - Chimie bio-organométallique: catalyse enzymatique», in Chimie organométallique et catalyse, EDP Sciences, 2020, p. 453‑470. doi: 10.1051/978-2-7598-1106-9.c024. |
| [39] | P. Morin, «Applications des lipases en synthèse énantiosélective», PhD Thesis, Citeseer, 2010. |
| [40] | M. Simard, La biocatalyse en synthèse énantiosélective: bioréduction de cétones à l’aide de tissus végétaux transformés et synthèse chimio-enzymatique et énantiosélective de la phosphonothrixine. Library and Archives Canada= Bibliothèque et Archives Canada, Ottawa, 2005. |
| [41] | E. C. Webb, Enzyme nomenclature 1992. Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes. Academic Press, 1992. |
| [42] | T. J. Colacot, «A Concise Update on the Applications of Chiral Ferrocenyl Phosphines in Homogeneous Catalysis Leading to Organic Synthesis», Chem. Rev., vol. 103, n° 8, p. 3101‑3118, août 2003, doi: 10.1021/cr000427o. |
| [43] | T. J. Colacot, «Ferrocenyl Phosphine Complexes of the Platinum Metals in Non-Chiral Catalysts», Platin. Met. Rev., vol. 45, no 1, p. 22‑30, 2001. |
| [44] | H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger, et R. M. Waymouth, «Stereospecific Olefin Polymerization with Chiral Metallocene Catalysts», Angew. Chem. Int. Ed. Engl., vol. 34, n° 11, p. 1143‑1170, juin 1995, doi: 10.1002/anie.199511431. |
| [45] | H. Sinn et W. Kaminsky, «Ziegler-Natta catalysis», in Advances in organometallic chemistry, vol. 18, Elsevier, 1980, p. 99‑149. |
| [46] | P. Dauban, L. Sanière, A. Tarrade, et R. H. Dodd, «Copper-Catalyzed Nitrogen Transfer Mediated by Iodosylbenzene PhIO», J. Am. Chem. Soc., vol. 123, n° 31, p. 7707‑7708, août 2001, doi: 10.1021/ja010968a. |
| [47] | H. Olivier-Bourbigou, «La catalyse biphasique», Actual. Chim., n° 5/6, p. 86‑90, 2002. |
| [48] | M. Hulce et D. W. Marks, «Organic-solvent-free phase-transfer oxidation of alcohols using hydrogen peroxide», J. Chem. Educ., vol. 78, n° 1, p. 66, 2001. |
| [49] | Mrs. G. Sabitha et A. V. S. Rao, «Synthesis of 3-Arylcoumarins, 2-Aroylbenzofurans and 3-Aryl-2H-1,4-benzoazines Under Phase-Transfer Catalysis Conditions», Synth. Commun., vol. 17, n° 3, p. 341‑354, févr. 1987, doi: 10.1080/00397918708077315. |
| [50] | T. Okawara, Y. Noguchi, T. Matsuda, et M. Furukawa, «Convenient syntheses of piperazine-2,5-diones and lactams from halocarboxamides using phase transfer catalysts», Chem. Lett., vol. 10, n° 2, p. 185‑188, févr. 1981, doi: 10.1246/cl.1981.185. |
| [51] | C. M. Starks, «Phase-transfer catalysis. I. Heterogeneous reactions involving anion transfer by quaternary ammonium and phosphonium salts», ACS Publications. [En ligne]. Disponible sur: https://pubs.acs.org/doi/pdf/10.1021/ja00730a033. |
| [52] | S. P. Neofotistos, A. Tzani, et A. Detsi, «Ionic Liquids: Advances and Applications in Phase Transfer Catalysis», Catalysts, vol. 13, n° 3, p. 474, févr. 2023, doi: 10.3390/catal13030474. |
| [53] | D. Zhao, Y. Wang, E. Duan, et J. Zhang, «Oxidation desulfurization of fuel using pyridinium-based ionic liquids as phase-transfer catalysts», Fuel Process. Technol., vol. 91, n° 12, p. 1803‑1806, déc. 2010, doi: 10.1016/j.fuproc.2010.08.001. |
| [54] | S.-S. Cheng et T. F. Yen, «Use of Ionic Liquids as Phase-Transfer Catalysis for Deep Oxygenative Desulfurization», Energy Fuels, vol. 22, n° 2, p. 1400‑1401, mars 2008, doi: 10.1021/ef700734x. |
| [55] | M. Nechab, «Synthèse et mise en oeuvre de nouveaux catalyseurs d’oxydation énantiosélectifs non métalliques», PhD Thesis, Université Joseph-Fourier-Grenoble I, 2006. |
| [56] | T. Pellegrini, «Asymmetric copper-catalyzed alkylations and autocatalysis», PhD Thesis, University of Groningen, 2019. |
| [57] | H. Wolf, «Organokatalysierte direkte asymmetrische a-Aminooxylierung von Aldehyden und Synthese von 2-Desoxy-D-erythro-pentose». |
| [58] | J. Seayad et B. List, «Asymmetric organocatalysis», Org. Biomol. Chem., vol. 3, n° 5, p. 719, 2005, doi: 10.1039/b415217b. |
| [59] | F. Capitta, «Utilisation des organocatalyseurs en synthese organique stereoselective», Consulté le: 19 décembre 2023. [En ligne]. Disponible sur: https://theses.hal.science/file/index/docid/807093/filename/VD_Annexes_CAPITTA_FRANCESCA_ 02042012.pdf. |
| [60] | B. Mokhtaria, Thèse de Doctorat synthèse originale organocatalyses de 1,2,3-Triazoles à partir d’Azides et de cetones non actives, 2012,6, Université d’Oran. |
| [61] | G. Lelais et D. W. MacMillan, «Modern strategies in organic catalysis: the advent and development of iminium activation», Aldrichimica Acta, vol. 39, n° 3, p. 79‑87, 2006. |
| [62] | C. Palomo et A. Mielgo, «Diarylprolinol ethers: Expanding the potential of enamine/iminium-ion catalysis», Angew. Chem.-Int. Ed. Engl.-, vol. 45, no 47, p. 7876, 2006. |
| [63] | A. Carlone, M. Marigo, C. North, A. Landa, et K. A. Jørgensen, «A simple asymmetric organocatalytic approach to optically active cyclohexenones», Chem. Commun., n° 47, p. 4928‑4930, 2006. |
| [64] | P. I. Dalko, «Do we need asymmetric organocatalysis?», Chimia, vol. 61, n° 5, p. 213‑213, 2007. |
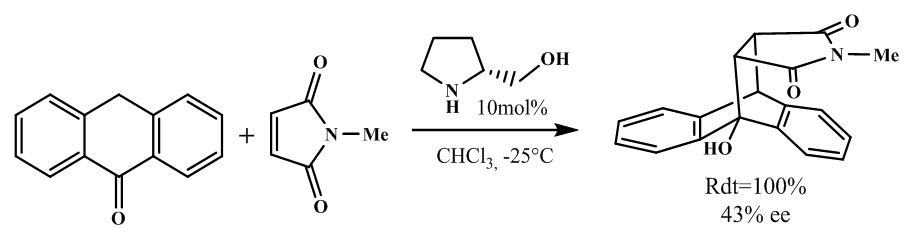
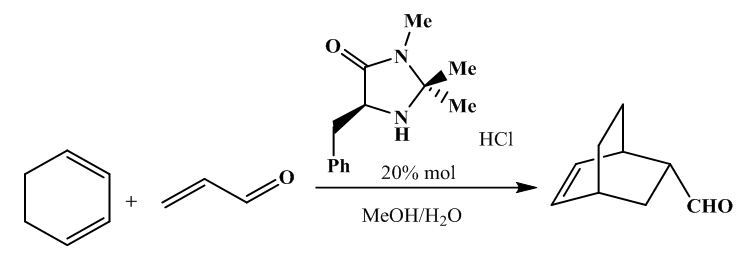
| [65] | M. Fofana, «Addition de michael énantiosélective organocatalysée de dérivés de nitrométhane sur les (e)-4-arylidènedihydrofurane-2,3-diones et (e)-1-benzyl-4-arylidènepyrrolidine-2,3-diones». 2022. |
| [66] | G. Stork, A. Brizzolara, H. Landesman, J. Szmuszkovicz, et R. Terrell, «The enamine alkylation and acylation of carbonyl compounds», J. Am. Chem. Soc., vol. 85, n° 2, p. 207‑222, 1963. |
| [67] | S. Mukherjee, J. W. Yang, S. Hoffmann, et B. List, «Asymmetric enamine catalysis», Chem. Rev., vol. 107, n° 12, p. 5471‑5569, 2007. |
| [68] | D. Seebach et al., «Are Oxazolidinones Really Unproductive, Parasitic Species in Proline Catalysis? – Thoughts and Experiments Pointing to an Alternative View», Helv. Chim. Acta, vol. 90, n° 3, p. 425‑471, mars 2007, doi: 10.1002/hlca.200790050. |
| [69] | M. C. Holland et R. Gilmour, «Deconstructing Covalent Organocatalysis», Angew. Chem. Int. Ed., vol. 54, n° 13, p. 3862‑3871, mars 2015, doi: 10.1002/anie.201409004. |
| [70] | A. Erkkilä, I. Majander, et P. M. Pihko, «Iminium catalysis», Chem. Rev., vol. 107, n° 12, p. 5416‑5470, 2007. |
| [71] | T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton, et D. W. MacMillan, «Enantioselective organocatalysis using SOMO activation», Science, vol. 316, n° 5824, p. 582‑585, 2007. |
| [72] | J. J. Devery, J. C. Conrad, D. W. C. MacMillan, et R. A. Flowers, «Mechanistic Complexity in Organo–SOMO Activation», Angew. Chem. Int. Ed., vol. 49, n° 35, p. 6106‑6110, août 2010, doi: 10.1002/anie.201001673. |
| [73] | M. Amatore, T. D. Beeson, S. P. Brown, et D. W. C. MacMillan, «Enantioselective Linchpin Catalysis by SOMO Catalysis: An Approach to the Asymmetric α-Chlorination of Aldehydes and Terminal Epoxide Formation», Angew. Chem., vol. 121, n° 28, p. 5223‑5226, juin 2009, doi: 10.1002/ange.200901855. |
| [74] | S. Rendler et D. W. C. MacMillan, «Enantioselective Polyene Cyclization via Organo-SOMO Catalysis | Journal of the American Chemical Society». Consulté le: 29 janvier 2024. [En ligne]. Disponible sur: https://pubs.acs.org/doi/abs/10.1021/ja100185p. |
| [75] | G. Forcher, «Vers la synthèse de carbènes N-hétérocycliques chiraux», These de doctorat, Le Mans, 2013. Consulté le: 19 février 2024. [En ligne]. Disponible sur: https://www.theses.fr/2013LEMA1030. |
| [76] | X. Bugaut et F. Glorius, «Organocatalytic umpolung: N-heterocyclic carbenes and beyond», Chem. Soc. Rev., p. 3511‑3522, 2011. |
| [77] | J. Pinaud, «Catalyse organique par les carbènes N-hétérocycliques (NHCs) et leur version supportée sur polymères à des fins de recyclage», These de doctorat, Bordeaux 1, 2010. Consulté le: 19 février 2024. [En ligne]. Disponible sur: https://www.theses.fr/2010BOR14135. |
| [78] | M. Wadamoto, E. M. Phillips, T. E. Reynolds, et K. A. Scheidt, «Enantioselective Synthesis of α,α-Disubstituted Cyclopentenes by an N -Heterocyclic Carbene-Catalyzed Desymmetrization of 1,3-Diketones», J. Am. Chem. Soc., vol. 129, n° 33, p. 10098‑10099, août 2007, doi: 10.1021/ja073987e. |
| [79] | B. Lygo et B. I. Andrews, «Asymmetric Phase-Transfer Catalysis Utilizing Chiral Quaternary Ammonium Salts: Asymmetric Alkylation of Glycine Imines», Acc. Chem. Res., vol. 37, no 8, p. 518‑525, août 2004, doi: 10.1021/ar030058t. |
| [80] | M. J. O’Donnell, «The preparation of optically active α-amino acids from the benzophenone imines of glycine derivatives», ChemInform, vol. 32, n° 38, p. no-no, 2001. |
| [81] | C. Nájera, «From α-Amino Acids to Peptides: All You Need for the Journey», Synlett, vol. 2002, n° 9, p. 1388‑1404, 2002, doi: 10.1055/s-2002-33552. |
| [82] | K. Maruoka et T. Ooi, «Enantioselective Amino Acid Synthesis by Chiral Phase-Transfer Catalysis», Chem. Rev., vol. 103, n° 8, p. 3013‑3028, août 2003, doi: 10.1021/cr020020e. |
| [83] | M. Z. Dimbi, M. Kapundu, E. Darimont, R. Warin, C. Delaude, et R. Huls, «Triterpentrïdes De Dodonaea Viscosa», Bull. Sociétés Chim. Belg., vol. 94, n° 2, p. 141‑148, 1985. |
| [84] | J. a. A. Ketelaar, «L’énergétique de la liaison hydrogène», J. Chim. Phys., vol. 46, p. 425‑428, 1949, doi: 10.1051/jcp/1949460425. |
| [85] | J.-M. Victor, «La structure de l’ADN en double hélice», Bibnum Textes Fond. Sci., 2012, Consulté le: 30 janvier 2024. [En ligne]. Disponible sur: https://journals.openedition.org/bibnum/503. |
| [86] | E. R. Jarvo et S. J. Miller, «Amino acids and peptides as asymmetric organocatalysts», Tetrahedron, vol. 58, no 13, p. 2481‑2495, 2002. |
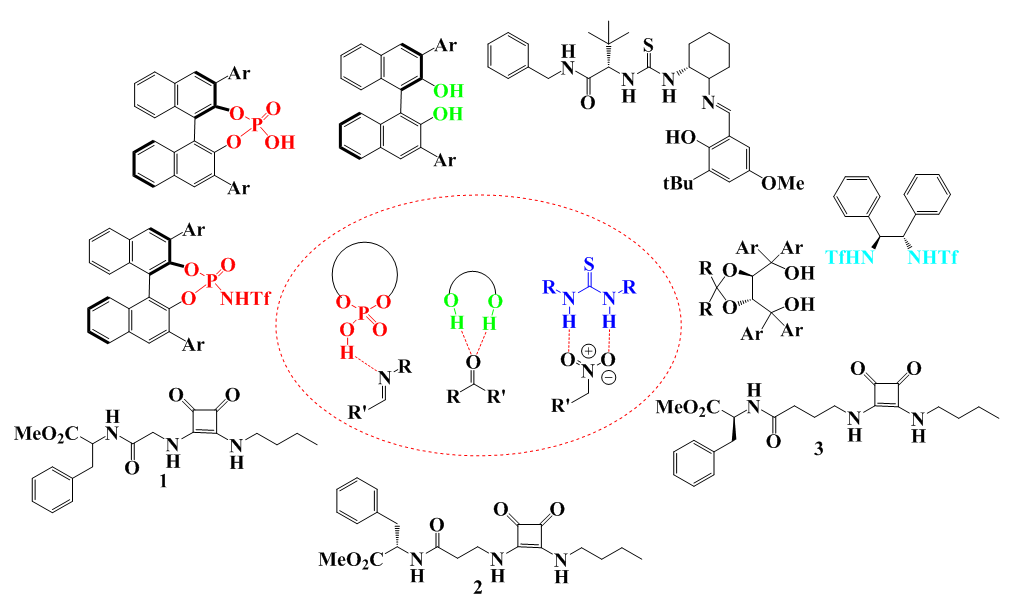
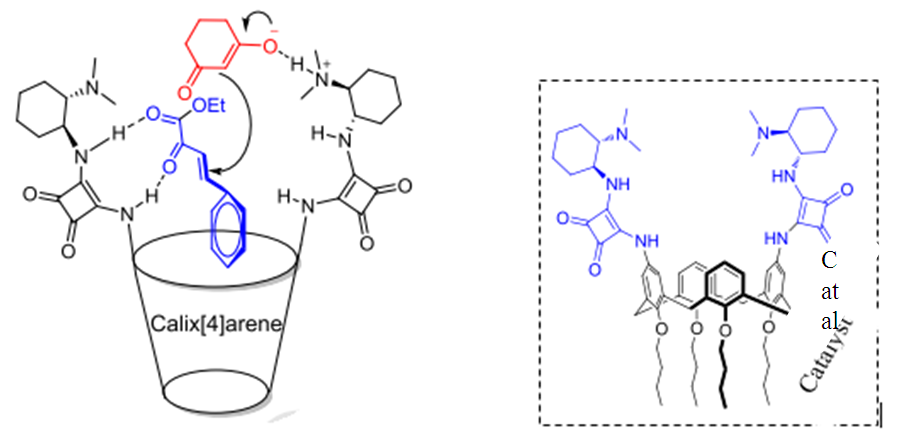
| [87] | U. Vural, M. Durmaz, et A. Sirit, «A novel calix [4] arene-based bifunctional squaramide organocatalyst for enantioselective Michael addition of acetylacetone to nitroolefins», Org. Chem. Front., vol. 3, n° 6, p. 730‑736, 2016. |
| [88] | K. Yang, Z. Ma, H.-X. Tong, X.-Q. Sun, X.-Y. Hu, et Z.-Y. Li, «Asymmetric Michael addition reactions catalyzed by a novel upper-rim functionalized calix [4] squaramide organocatalyst», Chin. Chem. Lett., vol. 31, n° 12, p. 3259‑3262, 2020. |
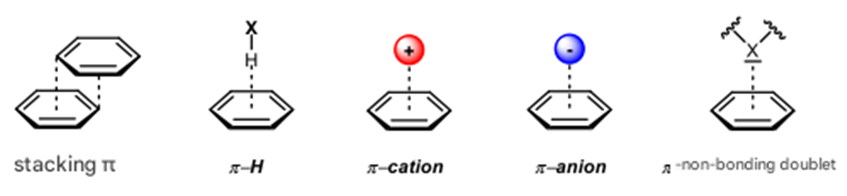
| [89] | G. Force, «Utilisation d’interactions non-covalentes pour le développement de nouvelles transformations», PhD Thesis, Université Paris-Saclay, 2021. Consulté le: 26 janvier 2024. [En ligne]. Disponible sur: https://theses.hal.science/tel-04055296/. |
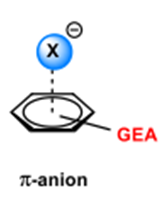
| [90] | D. Quiñonero et al., «Anion–π interactions: do they exist?», Angew. Chem., vol. 114, n° 18, p. 3539‑3542, sept. 2002, doi: 10.1002/1521-3757(20020916)114:18<3539::AID-ANGE3539>3.0.CO;2-M. |
| [91] | M. Egli et S. Sarkhel, «Lone Pair−Aromatic Interactions: To Stabilize or Not to Stabilize», Acc. Chem. Res., vol. 40, n° 3, p. 197‑205, mars 2007, doi: 10.1021/ar068174u. |
| [92] | T. J. Mooibroek, P. Gamez, et J. Reedijk, «Lone pair–π interactions: a new supramolecular bond?», CrystEngComm, vol. 10, n° 11, p. 1501‑1515, 2008. |
| [93] | A. J. Neel, M. J. Hilton, M. S. Sigman, et F. D. Toste, « Exploiting non-covalent π interactions for catalyst design », Nature, vol. 543, n° 7647, p. 637‑646, 2017. |
| [94] | L. Schuffenecker, G. Scacchi, B. Proust, J.-F. Foucaut, L. Martel, et M. Bouchy, «Thermodynamique et cinétique chimiques (collection Info-Chimie)», Librairie Lavoisier. Consulté le: 24 mars 2023. [En ligne]. Disponible sur: https://www.lavoisier.fr/livre/chimie/thermodynamique-et-cinetique-chimiques-collection-info-chimie/ schuffenecker/descriptif-9782852067196. |
| [95] | S. Selbmann, J. Herfurth, S. Petersen, et J. Ulrich, «Saccharose Inversion and Metastable Zone», Chem. Eng. Technol., vol. 38, n° 6, p. 1088‑1091, juin 2015, doi: 10.1002/ceat.201400684. |
| [96] | S. E. Barber, K. E. Dean, et A. J. Kirby, «A mechanism for efficient proton-transfer catalysis. Intramolecular general acid catalysis of the hydrolysis of 1-arylethyl ethers of salicylic acid», Can. J. Chem., vol. 77, n° 5‑6, p. 792‑801, juin 1999, doi: 10.1139/v99-080. |

 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML

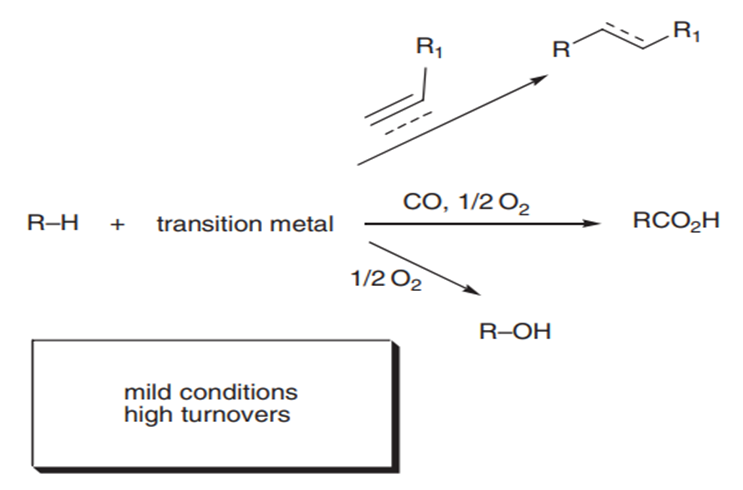
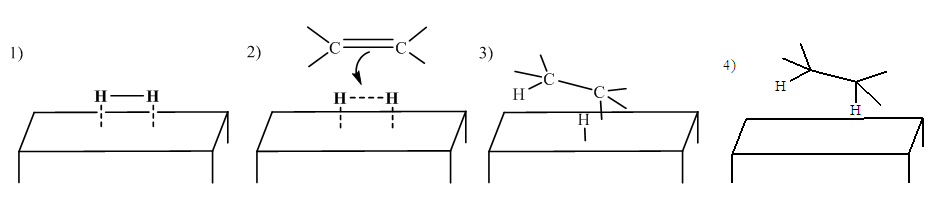
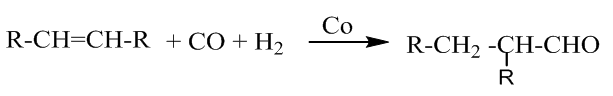

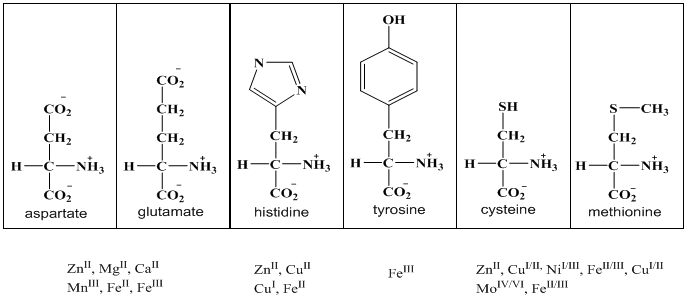
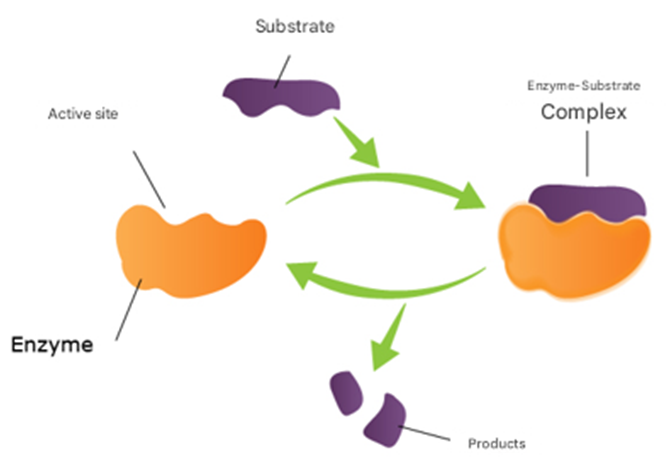
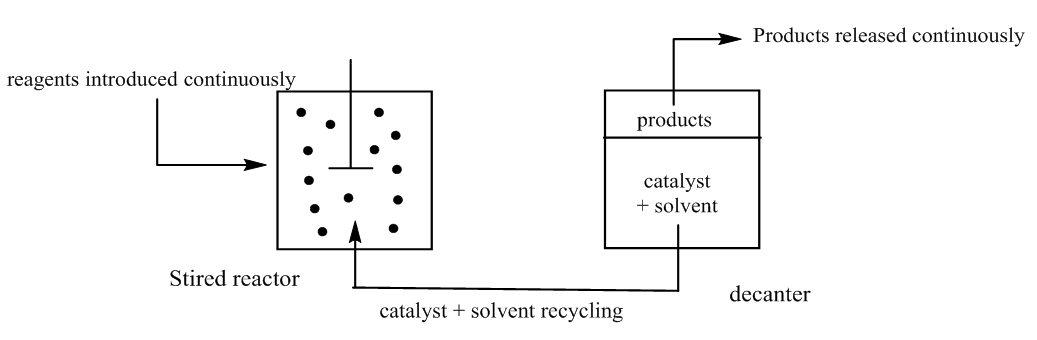
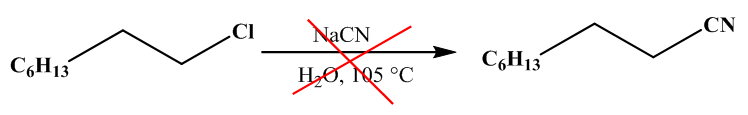
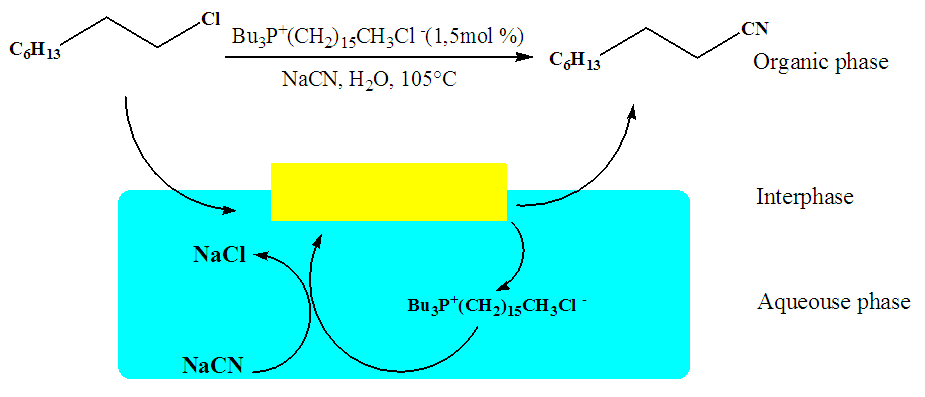
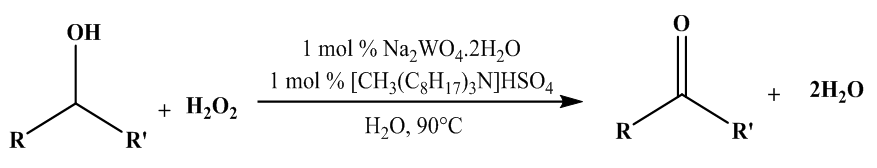
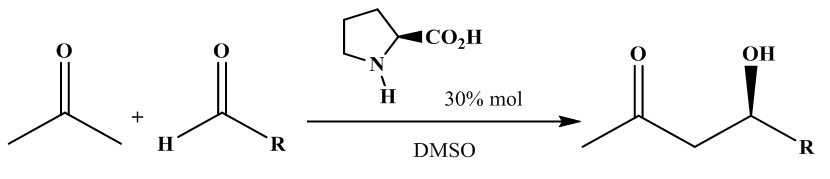
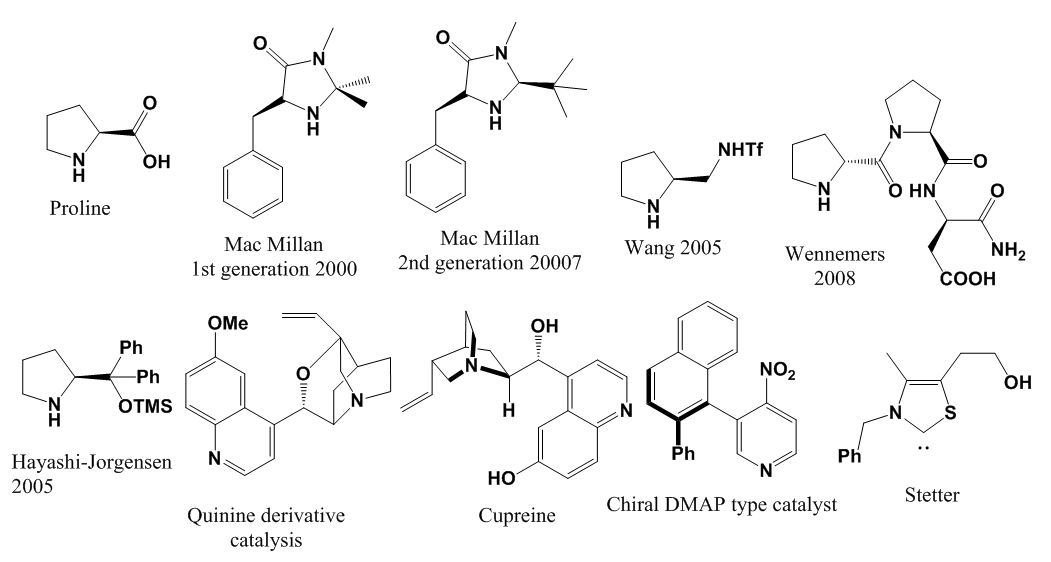
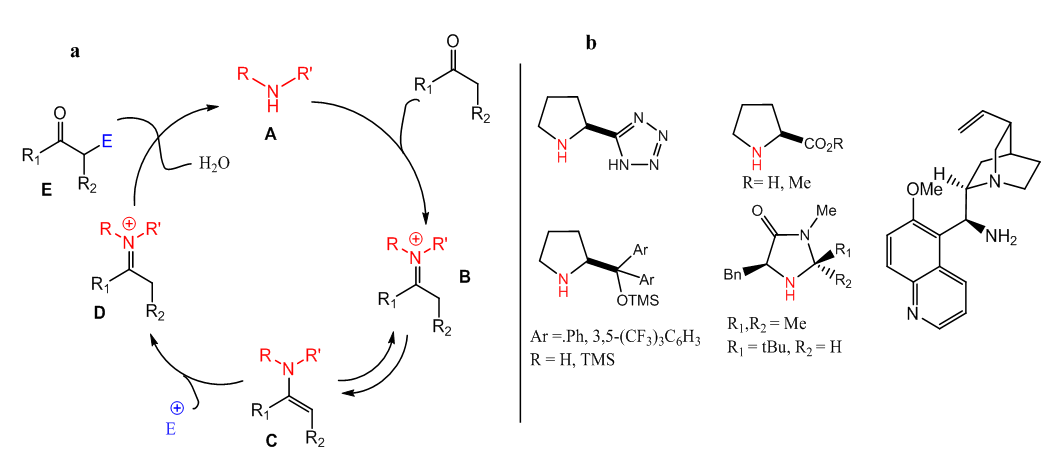
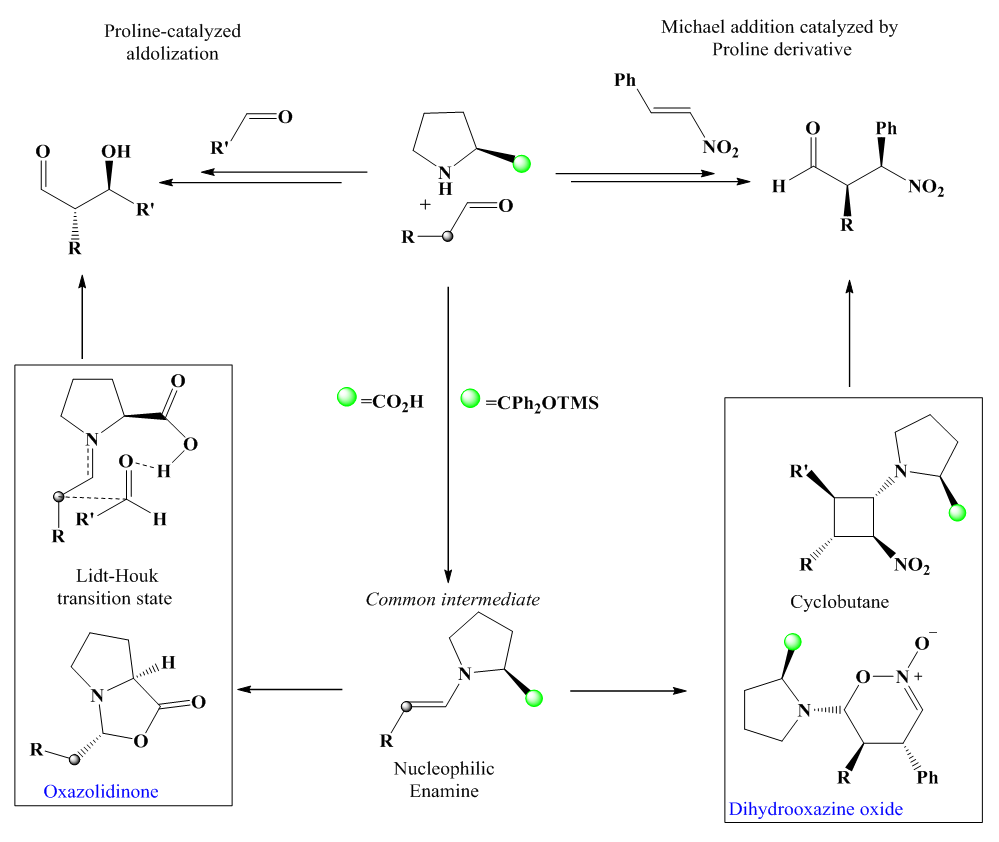
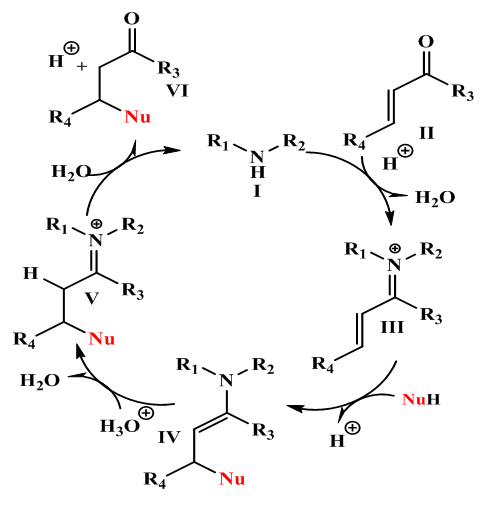
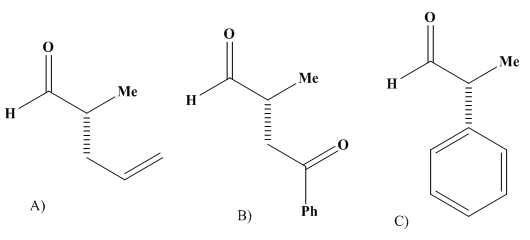
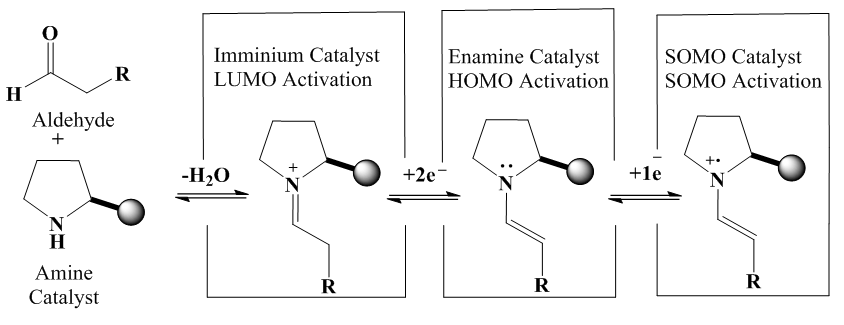
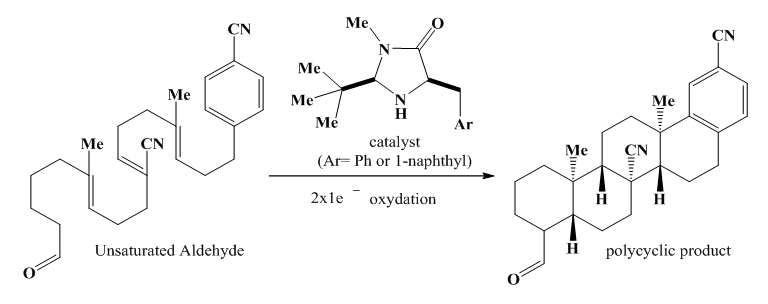
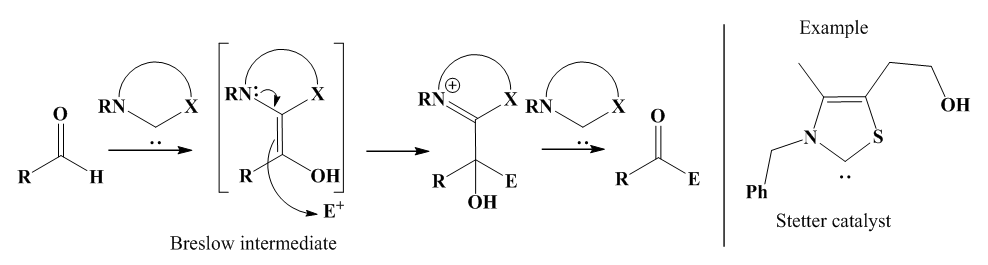
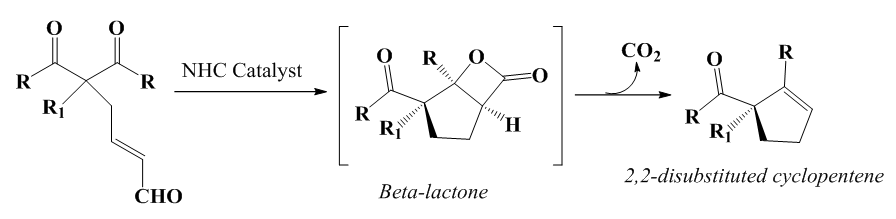
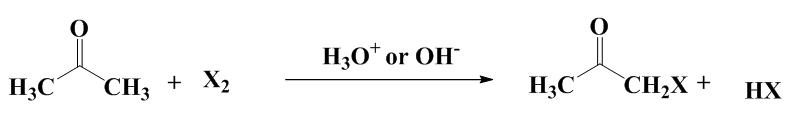
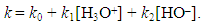 With k0, the rate constant of the uncatalyzed reaction. The value of k0 is very low compared to k1 and k2 (hence the notable effect of the increase in rate by the catalysts H3O+ and HO–). The addition of a weak acid only modifies the rate by the variation of the [H3O+] concentration that it allows, and not by the variation of its own concentration [95]. This indicates that it is specifically H3O+ the catalyst, and not just any acid.The inversion of sucrose is also of specific catalysis type. Its equation is:
With k0, the rate constant of the uncatalyzed reaction. The value of k0 is very low compared to k1 and k2 (hence the notable effect of the increase in rate by the catalysts H3O+ and HO–). The addition of a weak acid only modifies the rate by the variation of the [H3O+] concentration that it allows, and not by the variation of its own concentration [95]. This indicates that it is specifically H3O+ the catalyst, and not just any acid.The inversion of sucrose is also of specific catalysis type. Its equation is: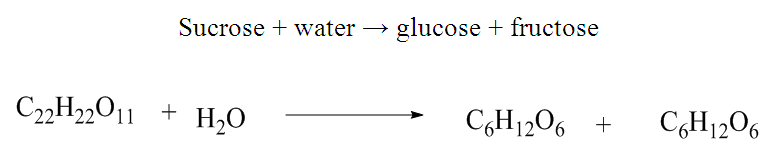
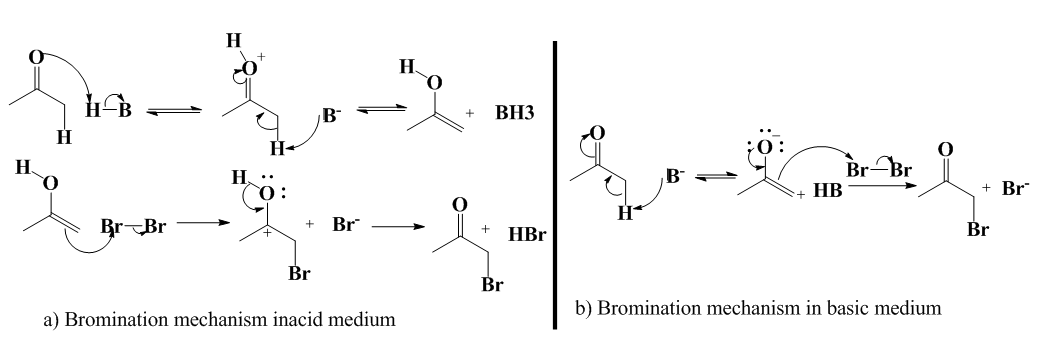
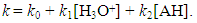 Where [AH] is the weak acid concentration.To demonstrate this property of general acid catalysis, it is necessary, for example, to determine the dependence of the rate (and therefore of k) on the amount of AH added, but this in a buffered medium, so that the term k1[H3O+] is kept constant. An example of general acid catalysis was studied by Barber et al. during a hydrolysis of tert-butyl ethers (1) and 1-arylethyl ethers (2) of salicylic acid [96]. This reaction is carried out with an efficient general acid catalysis by the -COOH group in ortho. The carboxylic group which catalyzes the reaction is intramolecular, so the hydrolysis is carried out by intramolecular hydrogen transfer. The mechanism is very different from classical general acid–base catalysis. Proton transfer occurs very rapidly in the strong hydrogen bond that develops and, although it is an integral part of the C-O bond cleavage process, there is virtually no coupling.
Where [AH] is the weak acid concentration.To demonstrate this property of general acid catalysis, it is necessary, for example, to determine the dependence of the rate (and therefore of k) on the amount of AH added, but this in a buffered medium, so that the term k1[H3O+] is kept constant. An example of general acid catalysis was studied by Barber et al. during a hydrolysis of tert-butyl ethers (1) and 1-arylethyl ethers (2) of salicylic acid [96]. This reaction is carried out with an efficient general acid catalysis by the -COOH group in ortho. The carboxylic group which catalyzes the reaction is intramolecular, so the hydrolysis is carried out by intramolecular hydrogen transfer. The mechanism is very different from classical general acid–base catalysis. Proton transfer occurs very rapidly in the strong hydrogen bond that develops and, although it is an integral part of the C-O bond cleavage process, there is virtually no coupling.