Ehouman Jean Missa1, Dembélé Georges Stéphane1, 2, Tuo Nanou Tiéba1, 3, Konaté Bibata1, 2, Soro Doh1, DJE Daniel Yannik1, Kodjo Charles Guillaume1, 4, Ziao Nahossé1, 2
1Laboratory of Thermodynamics and Physico-Chemistry of the Environment, Universite Nangui Abrogoua, Abidjan, Ivory Coast
2Ivorian Research Group on Disease Modelling (GIR2M), Universite Nangui Abrogoua, Abidjan, Ivory Coast
3Laboratory of Industrial Processes and Synthesis of the Environment and New Energies of the Institut Polytechnique FÉLIX HOUPHOUËT BOIGNY (INP-HB) of Yamoussoukro
4Laboratoire de Chimie BioOrganique et de Substances Naturelles, Université Nangui Abrogoua, Abidjan, Ivory Coast
Correspondence to: Dembélé Georges Stéphane, Laboratory of Thermodynamics and Physico-Chemistry of the Environment, Universite Nangui Abrogoua, Abidjan, Ivory Coast.
| Email: |  |
Copyright © 2024 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Abstract
The study of the stability and reactivity shows that compound B with a value of the energy gap of 3.749 eV is the most stable and the most electron accepting of the 2-(quinoline-4-yloxy)acetamide derivatives. Compound F, on the other hand, is the least stable and hardest of the series studied. The results of the indices of the dual descriptor obtained from the calculations at the theoretical level B3LYP in the base 6-31+G(d,p), showed that the C24 carbon atoms of the studied compounds are the most electrophilic sites. The C37 carbon atoms for compound A, C40 for compound B, C34 for compounds C and D and C33 for compounds E, F and G are the nucleophilic sites of the compounds.
Keywords:
Reactivity, DFT, Tuberculosis, 2-(quinoline-4-yloxy)acetamide
Cite this paper: Ehouman Jean Missa, Dembélé Georges Stéphane, Tuo Nanou Tiéba, Konaté Bibata, Soro Doh, DJE Daniel Yannik, Kodjo Charles Guillaume, Ziao Nahossé, Theoretical Study of the Chemical Reactivity of a Series of 2-(quinoline-4-yloxy)acetamide, American Journal of Chemistry, Vol. 14 No. 1, 2024, pp. 4-12. doi: 10.5923/j.chemistry.20241401.02.
1. Introduction
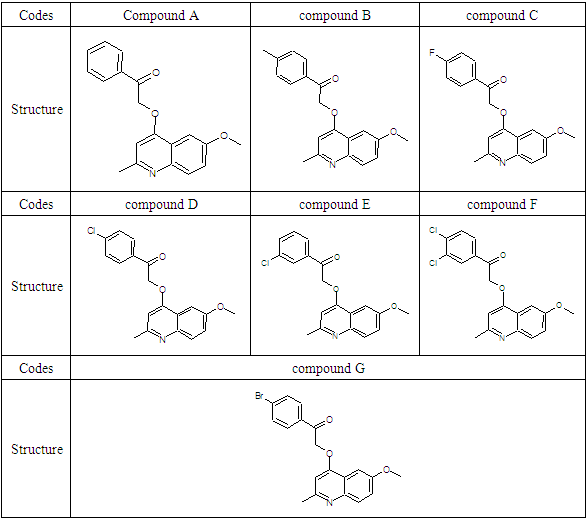
Tuberculosis is an infection caused by a bacterium (Mycobacterium tuberculosis) that most often affects the lungs. It is a preventable and treatable disease. Tuberculosis is spread from person to person through the air [1]. Tuberculosis remains a major infectious disease worldwide, with more than 10 million cases and 1.4 million deaths each year [2]. Today, for treatment, a combination of antibiotics and chemotherapy is used, but the treatment must be followed for at least 6 months (and up to two years in case of multi-resistant strains). Incomplete or poorly followed treatment is often responsible for the appearance of antibiotic-resistant tuberculosis which is then transmitted in the community. [2]. From these findings, there is an urgent need to find an effective treatment against tuberculosis. It is in this context that Borsoi et al. [3] synthesized a series of 2-(quinoline-4-yloxy)acetamide against M. tuberculosis H37Rv. Reactivity and stability are important studies in chemistry, as they provide insight into the chemical behavior of molecules. The general objective of this work is to evaluate the reactivity and stability of a series of 7 molecules derived from 2-(quinoline-4-yloxy)acetamide. The molecular structures of the studied molecules are presented in Table 1.Table 1. Structures of 2-(quinoline-4-yloxy)acetamide
 |
| |
|
2. Material and Method
2.1. Computational Level of Theory
In order to predict the sites of chemical reactivity of 2-(quinoline-4-yloxy)acetamide derivatives the quantum chemical calculations were performed using Gaussian 09 [4]. DFT methods are mainly used to generate the determination of reactivity sites in the studies. The geometric molecular optimization this work was conducted at the theoretical level B3LYP [5,6,7] in the 6-31+G(d,p) basis. This study was carried out by focusing on three theoretical approaches. The first one highlights the analysis of the electronic potential maps. The last approach is related to the boundary molecular orbitals. The third approach is related to local reactivity indices and dual descriptors. The hybrid functional confers the best energies and correlates with ab initio methods at high levels [8,9]. The geometries are kept constant for both cationic and anionic systems. The DFT conceptual model allows to obtain the global and local reactivity indices [10,11].
2.2. Reactivity Descriptors
2.2.1. Electrostatic Potential Surface Area
The molecular electrostatic potential (MEP) map is illustrated by the colors that vary from red to blue. The zero potential is represented by the green colored areas. It increases in the order red < orange < yellow < green < cyan < blue [12,13]. On the MEP map, the negative areas (red and yellow) of the MEP are electrophilic attack sites and the positive areas (cyan and blue) are nucleophilic attack sites.
2.2.2. Global Descriptors
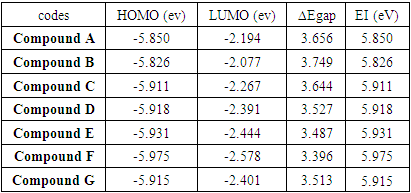
To predict the chemical reactivity, some theoretical descriptors related to the conceptual DFT have been determined. In particular, the energy of the lowest vacant molecular orbital (MO) (ELUMO), the energy of the highest occupied molecular orbital (MO) (EHOMO), the electronegativity (χ), the global softness (σ) and the global electrophilicity index (ω). These descriptors are all determined from the optimized molecules. It should be noted that, the descriptors related to the boundary molecular orbitals were calculated in a very simple way within the Koopmans approximation [14]. The LUMO energy characterizes the sensitivity of the molecule to a nucleophilic attack, and as for the HOMO energy, it characterizes the susceptibility of a molecule to an electrophilic attack. The electronegativity (χ) is the parameter which translates the aptitude of a molecule not to let escape its electrons. The overall softness (σ) expresses the resistance of a system to the change of its number of electrons. The global electrophilicity index characterizes the electrophilic power of the molecule. These different parameters are calculated from equations (1-6): | (1) |
 | (2) |
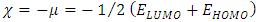 | (3) |
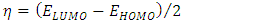 | (4) |
 | (5) |
 | (6) |
2.2.3. Local Descriptors and Dual Descriptors
The Fukui numbers of a molecule give information about the local reactivity in a molecule. The atom with the largest Fukui number is more reactive than the other atoms in the molecule [15]. These indices represent the qualitative description of the reactivity of atoms in the molecule. The Fukui function successfully predicts the relative reactivity for most chemical systems. The determination of Fukui indices for the selectivity of electrophilic and nucleophilic atoms in chalcone-derived compounds has been done. Ayers and Parr [16] explained that molecules tend to react where the Fukui function is largest when attacked by soft reagents and in places where the Fukui function is smallest when attacked by hard reagents. Using the Natural Atomic Population charges of the optimized ground state compounds, the Fukui function 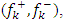 local softness
local softness 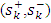 and local indices of electrophilia
and local indices of electrophilia 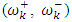 [17] have been determined. The Fukui functions are calculated using equations (7) and (8):
[17] have been determined. The Fukui functions are calculated using equations (7) and (8):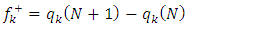 | (7) |
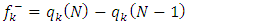 | (8) |
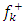 for nucleophilic attack
for nucleophilic attack 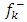 for electrophilic attack
for electrophilic attack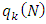 : Electronic population of atom k in the neutral molecule.
: Electronic population of atom k in the neutral molecule.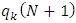 : Electronic population of atom k in the anionic molecule.
: Electronic population of atom k in the anionic molecule.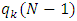 : Electronic population of atom k in the cationic molecule.The values of the dual descriptors [18,12] are obtained from equations (13 to 15)
: Electronic population of atom k in the cationic molecule.The values of the dual descriptors [18,12] are obtained from equations (13 to 15) | (9) |
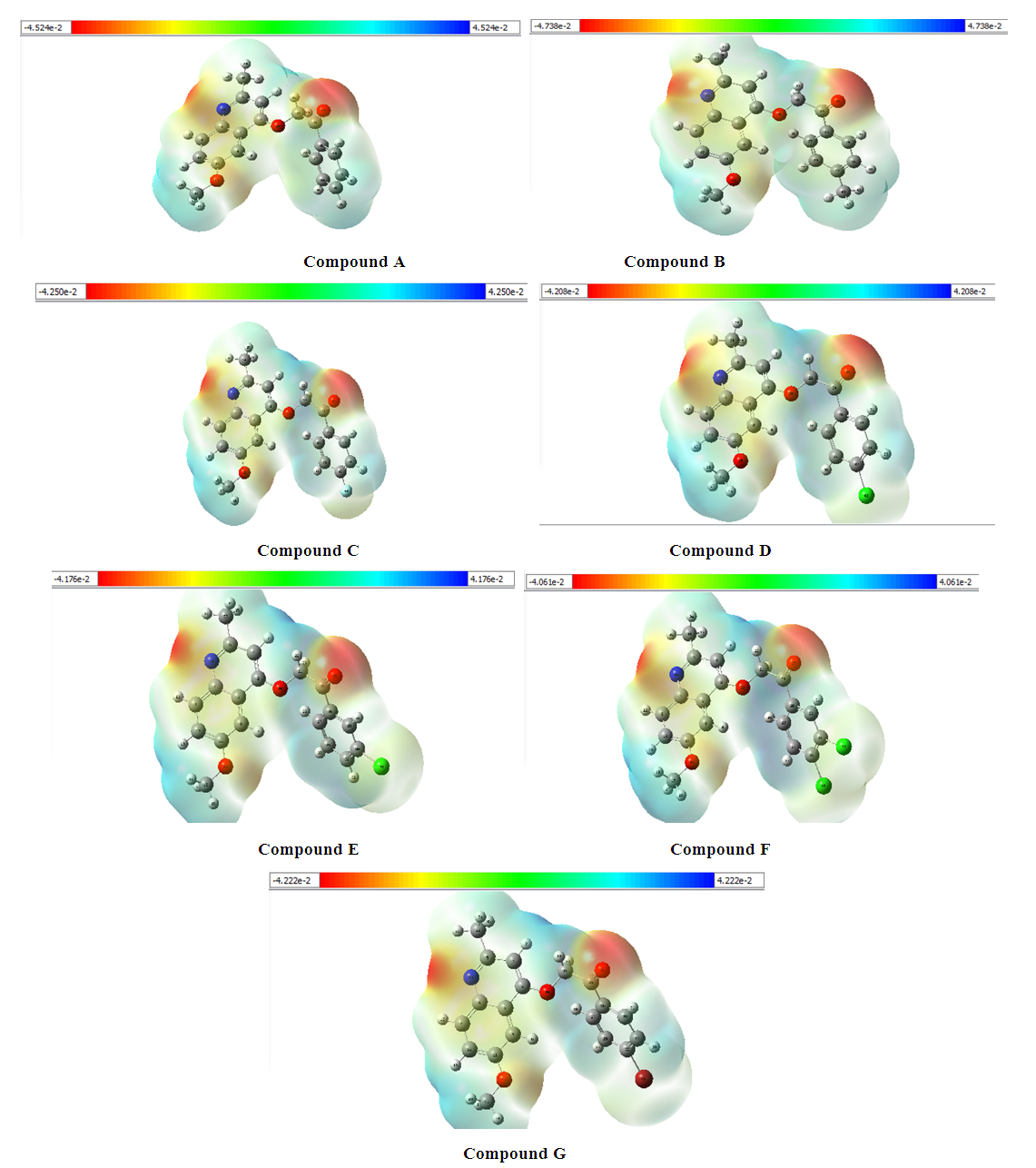 | Figure 1. Molecular Electrostatic Potential (MEP) surface of seven compounds |
2.3. Natural Population Analysis (NPA)
The calculation of natural atomic charge plays an important role in the study of molecular systems in quantum chemistry. For the quantitative description of a molecular charge distribution, the molecule is dissected into well-defined atomic fragments. A general and natural choice is to share the charge density at each point between the different atoms in proportion to their free atom densities at the corresponding distances of the nuclei [13]. In this work, the atomic charge values were obtained by the Natural population analysis.
3. Results and Discussion
3.1. Overall Reactivity
The results of table 2 highlight the values of the calculated energetic parameters. They show the compound B is the most stable with a value of the energy gap of 3.749 eV. Compound F having the most value of the energy gap (ΔEgap = 3.396 eV), it is the least stable of the seven studied compounds. Table 2. Energy parameters of the compounds studied
 |
| |
|
In Table 3 are listed the overall chemical reactivity indices. These overall reactivity results show that compound F has the smallest value of overall hardness (η = 1.698 eV). It is therefore the soft one of the series. Compound B on the other hand gives the largest value of electronegativity of (χ = 1.875 eV) and electrophilicity index (ω = 0.914 eV), this compound is more electron accepting of the series of 2-(quinoline-4-yloxy)acetamide derivatives.Table 3. Global descriptors of chemical reactivity
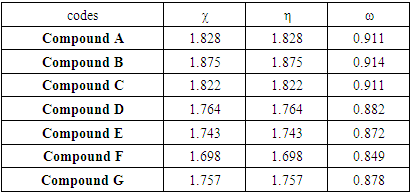 |
| |
|
3.2. Local Reactivity
3.2.1. Electrostatic Potential Surface
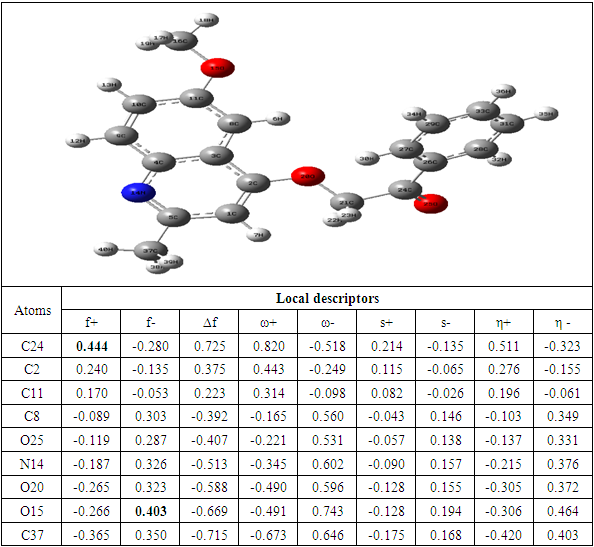
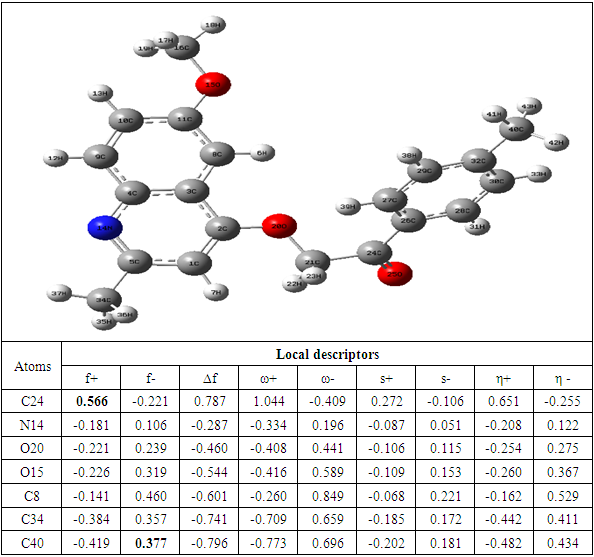
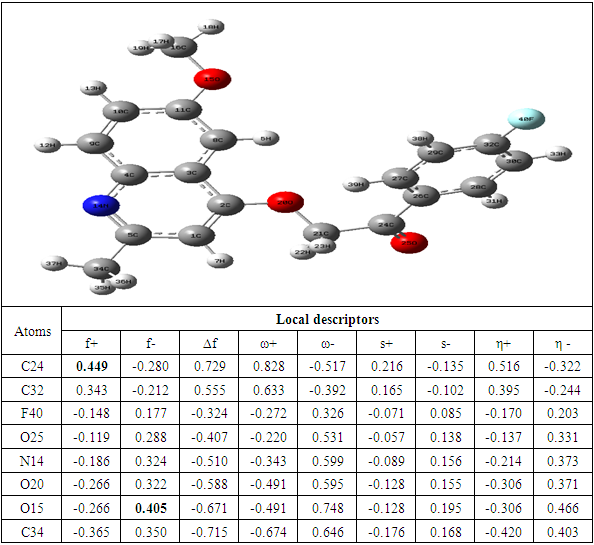
The determination of the molecular electrostatic potential (MEP) surfaces gives us a clear idea about the sites of molecular reactivity of the compounds in our study. The variation of the colors of this surface from red to blue allows to explain it better. The green areas have zero electrostatic potential, the yellow and red have negative potential and the cyan and blue have positive potential. The surface analysis shows that the C24 carbon atoms of the seven compounds of the 2-(quinoline-4-yloxy)acetamide series studied are electrophilic sites. As for the carbon atoms C37 for compound A, C40 for compound B, C34 for compounds C and D and C33 for compounds E, F and G, they were identified as nucleophilic sites. Carbon C24 is located in a negative electrostatic potential area and carbons C37, C40, C34 and C33 in the positive potential area.
3.2.2. Dual Reactivity Descriptors
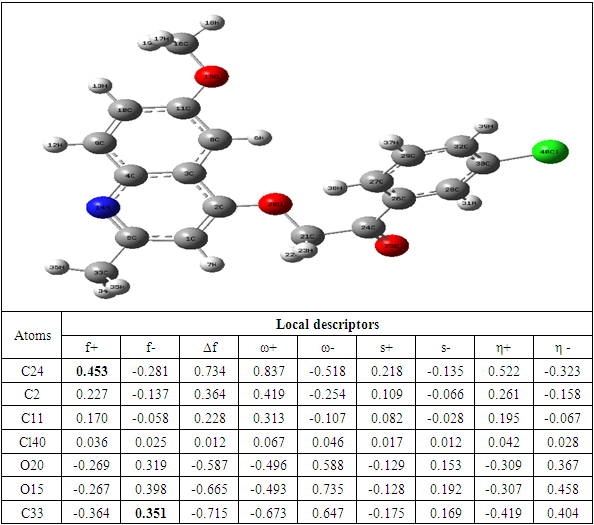
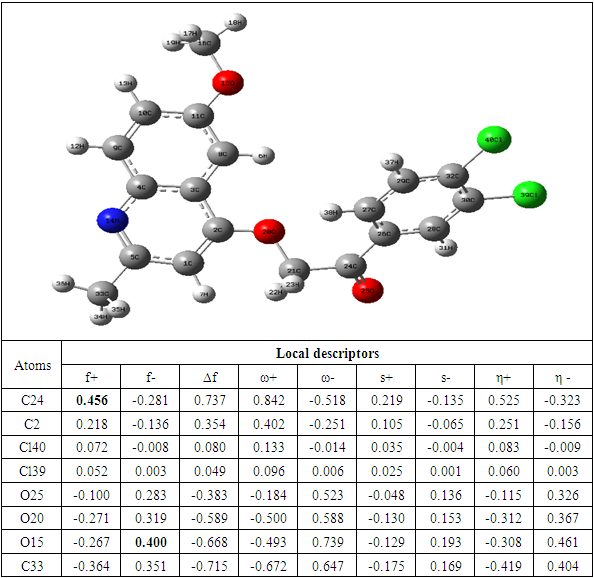
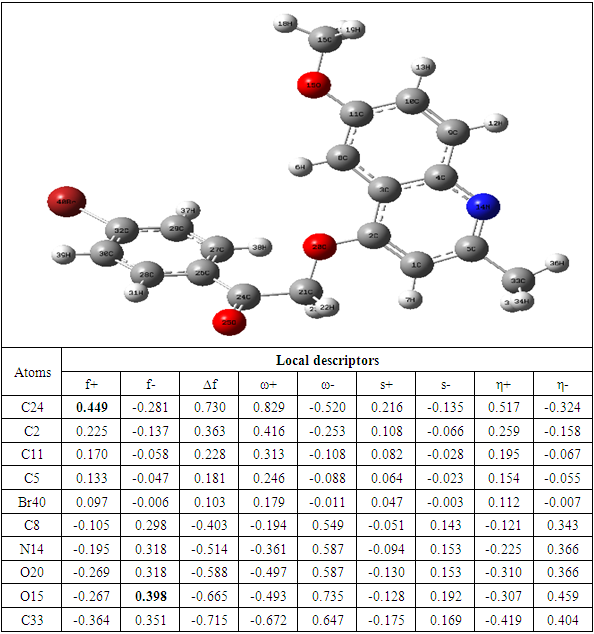
The theoretical level B3LYP/6-31+G (d, p) was used to determine the values of the dual descriptor of the different compounds. This analysis concerns only the atoms different from the hydrogen atom. The calculated dual descriptor values are recorded in Table 4 to 10. The results of the descriptor of the compound A grouped in table 4. They show that the carbon atom C24 is the favorable electrophilic site and the carbon C34 is the nucleophilic site.The results in Table 5 predict that the C24 carbon atom is the most favorable site for nucleophilic attack. With regard to the electrophilic attack site, the C40 carbon atom is the most favorable.The values of the various descriptors calculated indicate that the C24 carbon atom is the molecule's electrophilic site. For the nucleophilic site, the carbon atom C34 is the most privileged site.Table 7 predicts that the C24 carbon atom is the electrophilic site while the C34 carbon is the nucleophilic site.The identification of the reactive sites of compound E whose descriptor coefficient values are recorded in Table 8. These values obtained show that the carbon atom C24 is the most favorable electrophilic site and the carbon C33 is the most nucleophilic site.The results in Table 9 indicate that the most nucleophilic site is the C33 carbon atom. Carbon atom C24 is the most likely electrophilic site.The results in Table 10 indicate the most nucleophilic site is the C33 carbon atom. The C24 carbon atom is the most likely electrophilic site.Generally speaking, for the 7 compounds studied, the C24 carbon atom appears to be the most electrophilic.Table 4. Compound A reactivity descriptors calculated using natural population analysis (NPA)
 |
| |
|
Table 5. Compound B reactivity descriptors calculated using natural population analysis (NPA)
 |
| |
|
Table 6. Compound C reactivity descriptors calculated using natural population analysis (NPA)
 |
| |
|
Table 7. Compound D reactivity descriptors calculated using natural population analysis (NPA)
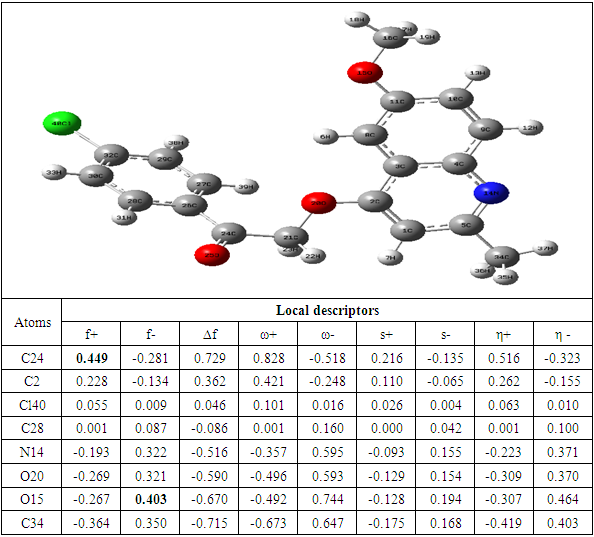 |
| |
|
Table 8. Compound E reactivity descriptors calculated using natural population analysis (NPA)
 |
| |
|
Table 9. Compound F reactivity descriptors calculated using natural population analysis (NPA)
 |
| |
|
Table 10. Compound G reactivity descriptors calculated using natural population analysis
 |
| |
|
4. Conclusions
The study of the stability and reactivity shows that compound B is the most stable and the most electron accepting of the derivatives used. On the other hand, compound F is the least stable and the least hard of the series studied. The analysis of the electrostatic potential surface and the results of the dual descriptor indices show that the C24 (largest value of the dual descriptor ∆f) carbon atoms of the studied compounds are identified as the more electrophilic sites. From the point of view of nucleophilic sites are the carbon atoms C37 for compound A, C40 for compound B, C34 for compounds C and D, and C33 for compounds E, F and G. As perspectives to this work, we plan to evaluate the reactivity of these compounds in different types of solvents, then to make a Natural Bonds Atomic study in order to see the electron displacements in the molecules and finally, to make a Molecular docking study.
References
| [1] | Organisation Mondiale de la Santé, "Tuberculose," 14 octobre 2021. [Online]. Available: https://www.who.int/fr/news-room/fact-sheets/detail/tuberculosis#:~:text=La%20tuberculose%20est%20 due%20%C3%A0,l'autre%20par%20voie%20a%C3%A9rienne.. [Accessed 25 March 2023]. |
| [2] | Institut pasteur, "Tuberculose," April 2021. [Online]. Available: https://www.pasteur.fr/fr/centre-medical/fiches-maladies/tuberculose. [Accessed 25 March 2023]. |
| [3] | A. Borsoi, J. Paz, B. Abbadi, F. Macchi, N. Sperotto, K. Pissinate, R. Rambo, A. Ramos, D. Machado, M. Viveiros, C. Bizarro, L. Basso and P. Machado, "Design, synthesis, and evaluation of new 2-(quinoline-4-yloxy)acetamide-based antituberculosis agents," Eur J Med Chem, vol. 15, no. 192, p. 112179, 2020. |
| [4] | M. J. Frisch, G. W. Trucks, H. B. Schlegel and G. E. Scuseria, "Gaussian 09, Revision A.02," Gaussian, Inc., Wallingford CT, 2009. |
| [5] | C. Lee, W. Yang and R. Parr., "Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density.," Physical Review Journals, vol. B37, p. 785, 1988.. |
| [6] | D. Becke, "Density-functional thermochemistry III. The role of exact exchange," Journal of Chemical Physics., vol. 98, p. 5648, 1993. |
| [7] | N. Tuo, G. Dembele, D. Soro, F. Konate, B. Konate, C. Kodjo and N. Ziao, "«Theoretical Study of the Chemical Reactivity of a Series of 2. 3-Dihydro-1H-Perimidine.," International Research Journal of Pure & Applied Chemistry, vol. 23, no. 11, pp. 13-25, 2022. |
| [8] | J. Kapp, M. Remko and P. v. R. Schleyer, "H2XO and (CH3)2XO Compounds (X= C. Si. Ge. Sn. Pb): Double bonds vs carbene-like structures can the metal compounds exist at all?," Journal of the American Chemical Society, vol. 118, pp. 5745-5751., 1996. |
| [9] | B. G. Johnson, P. M. Gill and J. A. Pople, "The performance of a family of density functional methods," The Journal of Chemical Physics, vol. 98, pp. 5612-5626., 1993. |
| [10] | R. G. Parr and R. G. Pearson, "Absolute Hardness: Companion Parameter to Absolute Electronegativity," J. Am. Chem. Soc., vol. 105, no. 26, p. 7512–7516, 1983. |
| [11] | W. K. Coulibaly, J. N’dri, M. G.-R. Koné, C. D. Dago, C. N. Ambeu, J.-P. Bazureau and N. Ziao, "Studies of the Chemical Reactivity of a Series of Rhodanine Derivatives by Approaches to Quantum Chemistry," Computational Molecular Bioscience, vol. 9, pp. 49-62, 2019. |
| [12] | J. S. N’dri, M.-R. Koné*, C. G. Kodjo, A. L. C. kablan, S. T. Affi, L. Ouattara and N. Ziao, "Theoretical Study of the Chemical Reactivity of Five Schiff Bases Derived From Dapsone by the DFT Method," Chemical Science International Journal, vol. 22, no. 4, pp. 1-11, 2018. |
| [13] | F. L. Hirshfeld, "Bonded-atom fragments for describing molecular charge densities," Theor. Chim. Acta, vol. 44, no. 2, p. 129–138, 1977. |
| [14] | T. Koopmans, "Úber die Zuordnung von Wellenfunktiomen und Eigenwerten zu den einzelnen Elektronen eines Atoms," Physica, vol. 1, pp. 104-113, 1934. |
| [15] | S. Dheivamalar, L. Sugi and K. Ambigai, "Density Functional Theory Study of Exohedral Carbon Atoms Effect on Electrophilicity of Nicotine: Comparative Analysis," p. 17–31, January 2016. |
| [16] | P. W. Ayers and R. G. Parr, "Variational Principles for Describing Chemical Reactions: The Fukui Function and Chemical Hardness Revisited," J. Am. Chem. Soc., vol. 122, no. 9, p. 2010–2018, Mars 2000. |
| [17] | K. Fukui, T. Yonezawa and H. Shingu, "A Molecular Orbital Theory of Reactivity in Aromatic Hydrocarbons," J. Chem. Phys., vol. 20, no. 4, p. 722–725, April 1952. |
| [18] | C. Morell, A. Grand and A. Toro-Labbé, "Theoretical support for using the Δf(r) descriptor," Chem. Phys. Lett, vol. 425, no. 4–6, p. 342–346, Juillet 2006. |

 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTML