-
Paper Information
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2023; 13(2): 42-48
doi:10.5923/j.chemistry.20231302.03
Received: Mar. 23, 2023; Accepted: Apr. 12, 2023; Published: Apr. 23, 2023

Forced Degradation in Pharmaceuticals – A Regulatory Update
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLSunita Sule1, Deepak Nikam1, Sushama Ambadekar1, Sudesh Bhure2
1Institute of Science, Analytical Chemistry Department, Madame Cama Road, Churchgate, Mumbai
2Chrom Specialities
Correspondence to: Sunita Sule, Institute of Science, Analytical Chemistry Department, Madame Cama Road, Churchgate, Mumbai.
| Email: |  |
Copyright © 2023 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Forced degradation study is essential to analytical method development and drug substance and product development. In the forced degradation study, the drug substance and drug product are subjected to external stress conditions, e.g., hydrolysis, oxidation, humidity, temperature and photolytic stress, and the stress's effect on the physicochemical stability of the material under study is accessed. Insights from the output of these studies help to ensure the safety, potency, and quality of the drug substance /drug product over its shelf life and in the development of stability indicating analytical method. This review article discusses the forced degradation process, its application, and the latest expectations from various regulatory guidance.
Keywords: Forced degradation, Mass balance, Peak purity, FDA guidelines, ICH guidelines
Cite this paper: Sunita Sule, Deepak Nikam, Sushama Ambadekar, Sudesh Bhure, Forced Degradation in Pharmaceuticals – A Regulatory Update, American Journal of Chemistry, Vol. 13 No. 2, 2023, pp. 42-48. doi: 10.5923/j.chemistry.20231302.03.
1. Introduction
- Impurities in a Drug substance (DS) or a Drug product (DP) can be generally classified as Organic (process, by-products, reagents, and degradation) impurities, Inorganic impurities (reagents, residual or heavy metals, inorganic salts.) and volatile impurities [1]. Some of the impurities are generated over a time due to interaction with excipients, packaging material or exposure to environmental conditions. Identification and qualification of these impurities is must to ensure the safety and stability of the drug substance and drug product. ICH Q3A (R2) and ICH Q3B (R2) guidelines provides the threshold for reporting, identifying, and quantifying the impurities observed in drug substance and drug products during real-time stability studies [1,2]. ICH has published specific guidance documents for conducting stability and photostability for drug substance and drug products [3,4]. Real-time stability studies take time. Hence, in order to know the potential degradation products, in the early days stage of product development, forced degradation studies are conducted by exposing the drug substance and drug product to different stress conditions to accelerate the degradation. Forced degradation study is very useful in finalising the analytical method as well as defining the specifications for impurities [5,6]. Methods developed using appropriate forced degradation study ensures acceptance of method for validation study and qualification in all method validation test parameters [7,8,9]. Various regulatory authorities such as USFDA, WHO, Japanese Health Authority (PMDA), Brazilian Drug regulatory agency (ANVISA) have published the specific recommendation for forced degradation and stress study [10,11,12,13,14]. Further, various literature on forced degradation are available in public domain [15,16,17]. The information available from literature is scattered and is limited to specific applications and regulations. This article simplifies and summarises the application, process and regulatory requirements for forced degradation studies.Advantages of forced degradation studies are manyfold. Applying the knowledge gained from forced degradation studies encompasses various aspects of the product life cycle. Major applications/benefits of forced degradation study are;- Identification of degradation pathways and intrinsic stability of Drug Substance and Drug products: Forced degradation data helps identify degradation pathways, e.g. sensitivity to temperature, humidity, oxidation, the nature of the observed impurity, i.e. process or degradation impurity and the chemical stability of the DS/DP under different stress conditions. These degradation impurities can be characterised and evaluated for their toxicity. - Drug substance development: FD study data can provide insights into the effect of selected reagents, solvents, salt form, and polymorph on chemical stability, providing input towards developing stable DS.- Formulation development: Identify drug/drug, drug/excipient, excipient/excipient, drug/drug product compatibility and aid selection of product component/s which would lead to developing a stable DP. - Product Stability: Provides in advance information on possible degradation impurities that could be formed during the product's shelf life under different environmental conditions and allow appropriate action. - Process Development: Information regarding the degradation pathways of DS/DP can be input in developing appropriate processes, e.g. alternate drying techniques like freeze drying instead of hot air ovens for heat-sensitive materials. - Define Handling and Storage Requirements: FD data indicating properties like Hygroscopicity and Photosensitivity of the DS/DP provides input for defining the manufacturing process, handling and storage condition of the product.- Define Product packaging: Provides input for selecting packaging material configuration, e.g. choice of the blister pack with appropriate moisture vapour transmission for packing of a hygroscopic Drug Product, and provides input in defining labelling requirements and transportation conditions.- Effect on safety and efficacy of product: Identification and studying the impact of degradation/ potential impurities which can develop or increase during shelf life on the safety and efficacy of DS/DP.- Development of analytical method: FD studies are an integral part of the validation of the analytical method. Impurities generated in FD studies aid in demonstrating specificity and stability-indicating properties of the analytical method.- Support robustness studies: Output of FD studies can provide relevant inputs for conducting robustness studies. - Support Setting up limits for impurities: the degradation data can be an input for setting specifications of degradation impurities identified in FD studies. - Support in investigations: Troubleshooting of OOS results during release / Stability testing. - Regulatory submissions: Forced degradation studies are a part of the regulatory submissions.
2. Forced Degradation Process
- Degradation Products: Drug substances and drug products can undergo degradation due to chemical instability or chemical interactions with other formulation components, leachable or interaction with immediate packing material, or are developed due to exposure to physical stress during the manufacturing process, which results in the development of undesired moiety. The effect of incompatible environmental conditions like loss of volatile components or degradation of a thermolabile component due to temperature, degradation, or microbial degradation due to humidity, light during manufacturing, and storage can render the drug product degraded. Degradation may result in the loss of the potency or compromise appearance or desired physical attributes, e.g. viscosity, hardness, particle size or change in a polymorph of the DS/DP, adversely affecting the quality of the product. Sometimes, the degradation may result in the formation of impurities leading to unwanted pharmacological or toxicological effects affecting patient safety. Thus these impurities need to be identified, characterised and controlled.As per ICH Q1A, product stability needs to be studied under three conditions – Accelerated, Intermediate and Controlled Room temperature to identify the impurities generated due to the degradation the DS/DP undergoes during its shelf life. This information inputs into the decision-making for the shelf life of a DS/DP. In forced degradation studies, the DS/DP are deliberately exposed to different environmental stress conditions, e.g. Hydrolysis, Oxidation, Humidity, Temperature and Photolytic stress. The reaction to the stress, i.e. degradation of the drug and generation of impurities is studied. Currently, there are no clear guidelines on the amount of degradation to be achieved. However, the industry-accepted range is between 5 – 20% of degradation [15,16,17]. The stress level should be above the accelerated condition. The rationale being the degradation should be enough to generate primary degradants. However, caution must be applied so that unrealistic high degradation resulting in secondary degradation does not take place. The placebo of the drug product shall be exposed to similar stress. It is essential to ensure that only degradation peaks due to DP are considered, and degradation peaks due to placebo are not considered. It is beneficial to conduct forced degradation studies during the product development stage to gather insights into the stability and degradation pathway to arrive at the final optimised DS/DP configuration and stability indicating analytical method. Once the final form and synthesis route are selected for DS, a confirmatory study is done. While for DP, the confirmatory study is done once the formulation and packaging are finalised. Confirmatory FD studies are often included in regulatory submissions. For DS photostability data of both FD and Confirmatory studies are a part of the submission documents. Stress conditions Stress conditions for DS / DP are to be selected to achieve sufficient degradation between 5-20%. Wherever possible, sample should be in a dissolved state. Samples solutions can be kept over time to achieve the desired degradation or refluxed to accelerate degradation. The time of exposure may vary to achieve the desired degradation. The concentration of the degrading solution can also be adjusted to aid the desired level of degradation. Some DS/ DP may be highly stable and may not exhibit any degradation for selected degradation conditions, in such cases, records for the degradation trials with unsuccessful efforts shall be maintained. A general outline for process of conducting the stress studies is outlined in Table 1, the typical exposure conditions for DS/ DP are reported in Table 2 and the samples to be exposed are reported in Table 3.
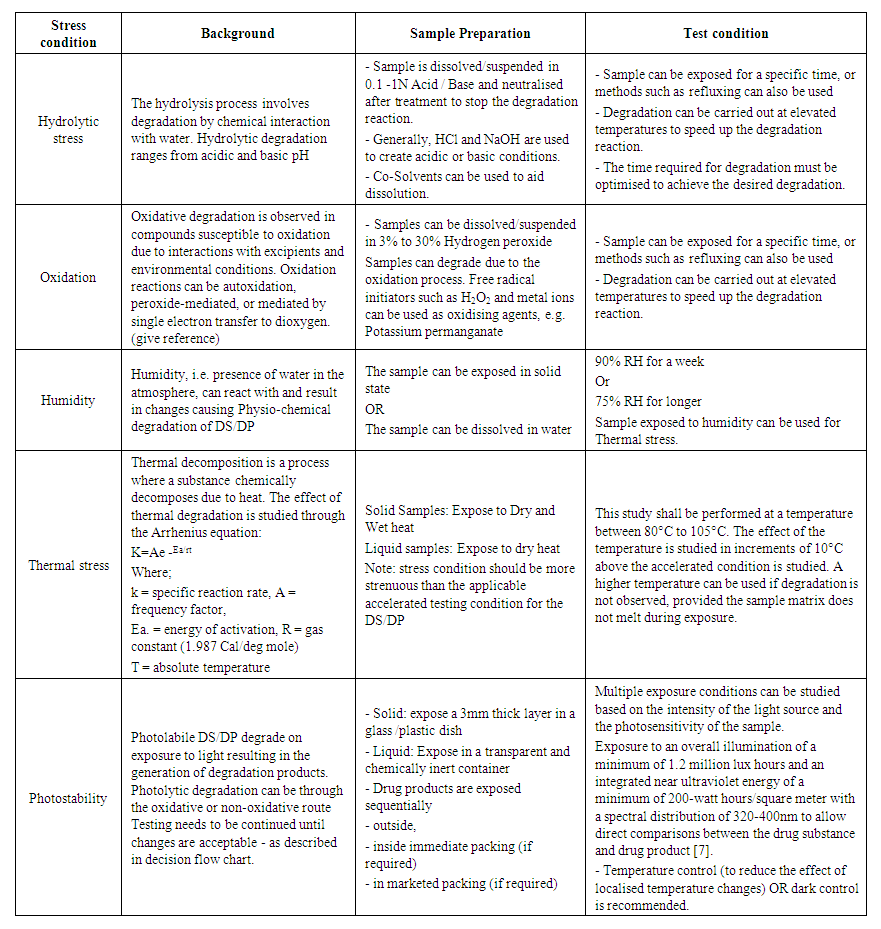 | Table 1. Stress Test Conditions |
|
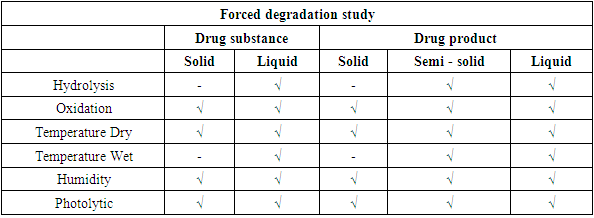
|
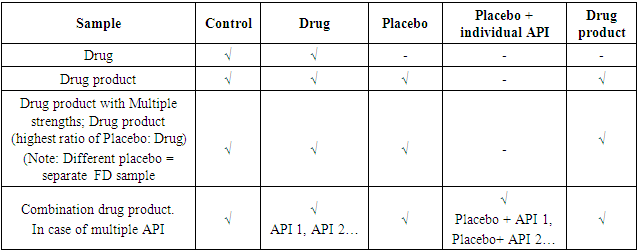
3. Regulatory
- Although forced degradation studies are conducted during product development, most regulatory bodies expect confirmatory forced degradation studies to be a part of the submission process for novel and generic pharmaceutical formulations. As the pharma industry develops more complex/new molecules and formulation types, the onus is on the analytical R&D to find the most suitable approach to conduct forced degradation studies. Most of the guidelines mentioned below provide a general outline on forced degradation studies other than ICH Q1B which specifically addresses the photostability studies and Anvisa RDC 53 which gives detailed regulatory requirements for FD studies. ICH Q1 A (R2): Stability testing of new drug substances and products [3]This guideline proposes a method of stress testing in Section 2.1.1. Stress testing. It recommends that stress testing should be carried out on a single batch of drug substances and should study; - effect of temperature (in increments of 10 deg. C, above that for Acc. testing)- humidity (75% RH or above)- hydrolysis (over a wide range of pH in case of solution or suspension)- oxidation (where appropriate)- photolysis (where appropriate, as per ICH Q1B) Degradation products not formed during accelerated or long-term stability need not be monitored. The Stress study data should be a part of the data package submitted to regulatory authorities. Further, as per section 2.2.4 Container closure system, Stress testing of the drug product outside its immediate container or other packing material can be an input to stress testing of the dosage form.ICH Q1B: Stability testing: Photostability testing of new drug substances and products [4] This guideline recommends samples, test conditions and data evaluation for photostability studies. In case of drug substance, testing is recommended to be conducted on one batch per part as follows;Part 1 - Forced degradation testing (conditions can be harsher than confirmatory testing)Part 2 – Confirmatory testingIn the case of Drug Products, photostability testing is done on one batch during the development phase and on one confirmatory batch. The testing is conducted in a sequential manner as follows;- Exposed drug product outside immediate packing- If necessary, Expose drug products in the immediate packing- If necessary, Expose drug products inside the marketing packingTesting products like infusions and creams to support in-use photostability is also recommended.ICH Q2 (R1): Validation of analytical procedures: text and methodology [7]ICH Q2 R1 provides guidance on the execution of method validation and the various parameters to be included. Section 1.2 deals with the Specificity section of method validation. The guidance proposes that specificity can be demonstrated by spiking DS/DP with an appropriate level of impurities when impurities are available or in case the impurities are unavailable by comparing the test results with another validated methods. The guidance recommends using samples stressed for Light, Heat, Humidity, Hydrolysis, and oxidation conditions. ICH Q3 A (R2): Impurities in New Drug Substances [1] This guidance discusses the classification of impurities, their origin and the rationale for reporting and controlling those impurities. Section 3.1 discussed the rationale for reporting organic impurities, and the result of the stress test conducted as per ICH Q1 A are to be summarised and submitted by the applicant.ICH Q3 B (R2): Impurities in New Drug Products [2] This guidance refers to the impurities in the new drug product, specifically degradation products of the DS or impurities arising out of the interaction of DS with excipients or interaction of DS with immediate container closure systems. Section 3 of the guidance refers to the capability of Analytical procedure for detection and content and qualification of quantification of degradation products. The analytical method should be validated with the demonstration of specificity for specified and unspecified degradation impurities, which have been studied under stress conditions like light, heat, humidity, acid/base hydrolysis, and oxidation. The degradation product can be quantified against a known impurity standard or DS concentration using an appropriate response factor. Reporting threshold, Identification Threshold, and Qualification threshold limit are calculated on the maximum daily dose of the DS administered per day. Further, the reporting of degradation products should include Specified Identified, Specified unidentified, Unspecified degradation product ≤ identification threshold and Total degradation product.ICH M4Q (R1): Common Technical Document for the Registration of Pharmaceuticals for Human Use – Quality [5] This guidance provides, in detail, a description of the tests, studies, and documents to be submitted for a DS /DP registration. Module 3.2.S.7 refers to stress studies and mentions that the results from the stress studies and forced degradation studies need to be summarised and presented as tabular, graphic or narrative. The conclusion based on the outcome of these studies concerning storage conditions/shelf life/retest date must also be summarised. Further, information on the analytical method used for these studies and their validation must also be submitted.FDA Guidance: Analytical procedures and method validation for drugs and Biologics [9]This guidance provides recommendations for submitting analytical procedures and validation data for DS/DP covered under NDA, ANDA and BLA. It encompasses information on analytical method development, validation and life cycle management of analytical procedures. It also discusses the content of analytical procedures, reference standard and materials. In sec 4, analytical method validation, reference is made to the samples undergoing exposure to stress conditions to demonstrate specificity. If an analytical method is transferred to another lab, it is stability indicating method. It is recommended that stressed samples be analysed at both sites. FDA Guidance: Guidance for Industry ANDAs: Stability Testing of Drug Substances and Products [10]In this guidance, the FDA recommends that the applicant follow ICH stability guidance Q1 A, Q1 B, Q1 C, Q1 D and Q1 E for submission batches. Thus, the stress studies must be performed as mentioned in Q1A and Q1B. If the applicant follows an alternate approach, they must justify it to the FDA. US Pharmacopoeia: Validation of compendial Procedures <1225> [8]In case the degradant or contaminant standard is unavailable, this guidance recommends testing degradation products/contaminants using an alternate analytical procedure under the same accelerated condition to demonstrate the specificity of the method. WHO technical report series; Annex 10 Stability testing of active pharmaceutical ingredients and finished pharmaceutical products [11]This guidance refers to the stability testing and documentation for submission of Active pharmaceutical ingredients (API), Finished pharmaceutical products (FPP), and Biologics.As per Section 2.1.2. Stress testing needs to be carried out on one batch of API and should study - effect of temperature (in increments of 10 deg. C above temperature used for accelerated testing)- humidity (at a level above the accelerated condition for the DS / DP)- hydrolysis (over a range of pH in case of solution or suspension)- oxidation- photostability testing Degradation products not formed during accelerated or long-term stability need not be monitored.These studies' results form part of the data package submitted to regulatory authorities. Alternatively, submitting the relevant data regarding identifying degradation products and pathways is acceptable if available and published in the scientific literature. Section 2.2.2 refers to the stress testing of FPP; Photostability testing needs to be conducted on one primary batch, and other stress tests are to be conducted as mentioned in ICH guidelines. Further, Section 2.2.4 Container closure system recommends; Stress testing of the drug product outside its immediate container or other packing material can be input to stress testing of the dosage form. Formulation-specific studies are also recommended, e.g. freeze-thaw cycle for liquid products and cyclic studies for semi-solids.Japanese Pharmacopoeia [12]The proposed analytical method should be specific and capable of identifying and estimating the analyte. If the reference standards for impurities are unavailable for comparative studies, degradation products from the stressed condition may be used for further studies. ANVISA RDC 53, 2015 [13]This document guides the process for conducting FD studies, identification and qualification of degradation products and documentation for submission. FD studies should be conducted on a laboratory, pilot or commercial scale batch. The product, placebo and active pharmaceutical ingredient in the formulation need to be subjected to stress. In case of multiple strength- all concentrations and in case of fixed combination, individual API and formulation must be subjected to the stress. The FD conditions include exposure to Heating, Humidity, Acid solution, Basic Solution, Oxidizing solution, Photolytic conditions and a new stress condition metal ion. Justification needs to be given in case FD is not performed in any condition. Exposure should be such that a minimum of 10% Degradation is observed and justified if not achieved. Evidence of peak purity demonstrates no interference from formulation components, impurities and degradants. FD data should be input into developing analytical methods. FD studies need to be redone for API in case of change in synthesis route, API from each supplier and when there are qualitative or quantitative changes in excipient composition. This guideline lays down requirements and limits notification, identification and qualification of degradation products in DS/DP. The acceptance limit for individual and total degradation products must be input in release and stability specifications. ANVISA RDC 166, 2017 [14]Chapter IV, section 1 deals with the Selectivity analytical method. As per Art 21, it is required that for quantitative determinations and limit tests, selectivity should be demonstrated by proving that the peak obtained is due to analyte and is free from the interference of diluent, matrix, impurities, and degradants. This must be supported by testing samples exposed to degradation conditions with a wide range of pH, oxidation, heat, and light. For comparison, the execution of the study must also be carried out with the formulation, with the placebo and in the isolated and associated active pharmaceutical ingredient(s) in the case of dose associations fixed. The study of the forced degradation. profile must be carried out at all drug concentrations. In the existence of more than one manufacturer of the active pharmaceutical ingredient, the results of forced degradation must be evaluated for each manufacturer.
4. Conclusions
- Forced degradation studies designed for selected pharmaceutical products provides deep insight into the potential degradation impurities during product development. The broad guidelines published by various regulatory authority provides a general outline for forced degradation studies. However, the expectation from all the regulatory authorities is to have a detailed forced degradation study to understand of degradation pathway for the impurities and to establish the identity of each major degradation impurity. Understanding of degradation pathway enables the development of stability-indicating analytical methods and prediction of product quality, stability and shelf life. In addition, knowledge gained from the forced degradation study provides insight into safety, selection of excipients, handling process, manufacturing process, packaging, storage conditions and labelling for the drug substance/drug product.