-
Paper Information
- Previous Paper
- Paper Submission
-
Journal Information
- About This Journal
- Editorial Board
- Current Issue
- Archive
- Author Guidelines
- Contact Us
American Journal of Chemistry
p-ISSN: 2165-8749 e-ISSN: 2165-8781
2023; 13(1): 26-31
doi:10.5923/j.chemistry.20231301.03
Received: Dec. 25, 2022; Accepted: Jan. 5, 2023; Published: Jan. 13, 2023

Synthesis of Bimetallic (Fe-Re, Fe-Mn) and Triosmium Carbonyl Complexes Containing Bromine, Nitrogen, Sulfur and Phosphorus Donor Ligands
 Abstract
Abstract Reference
Reference Full-Text PDF
Full-Text PDF Full-text HTML
Full-text HTMLArjun Chandra Bhowmick1, 2, 3, Shishir Ghosh3, Shariff Enamul Kabir3
1Mawlana Bhashani Science and Technology University, Santosh, Tangail, Bangladesh
2Kalamazoo College, Department of Chemistry and Biochemistry, Kalamazoo, Michigan, USA
3Department of Chemistry, Jahangirnagar University, Savar, Dhaka, Bangladesh
Correspondence to: Arjun Chandra Bhowmick, Mawlana Bhashani Science and Technology University, Santosh, Tangail, Bangladesh.
| Email: |  |
Copyright © 2023 The Author(s). Published by Scientific & Academic Publishing.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/

Reaction of [Fe2(CO)8(κ2-dppm)] (1) with [Re2(CO)6(µ-κ3-pyS)2] (2) in refluxing toluene affords [FeRe(CO)7(κ2-pyS)(κ2-dppm)] (3) a new mixed-metal complex. Similarly, refluxing the complex [Fe2(CO)8(κ2-dppm)] (1) with [Re4(CO)12(µ-κ3-pymS)4] (4) in tolueneform the isostructural complex [FeRe(CO)7(κ2-pymS)(κ2-dppm)] (5). Again, Treatment of electronically unsaturated cluster [(µ-H)Os3(CO)8(Ph2PCH2P(Ph)C6H4)] (6) with HBr gas at ambient temperature in dichloromethane afforded a complex [(µ-H)Os3(CO)8(µ-Br)(µ-dppm)] (7). All complexes have been characterized spectroscopically. Additionally, complexes 1 and 7 have been characterized by single-crystal X-ray diffraction studies.
Keywords: Rhenium, Iron, Manganese, Osmium, Bromide
Cite this paper: Arjun Chandra Bhowmick, Shishir Ghosh, Shariff Enamul Kabir, Synthesis of Bimetallic (Fe-Re, Fe-Mn) and Triosmium Carbonyl Complexes Containing Bromine, Nitrogen, Sulfur and Phosphorus Donor Ligands, American Journal of Chemistry, Vol. 13 No. 1, 2023, pp. 26-31. doi: 10.5923/j.chemistry.20231301.03.
Article Outline
1. Introduction
- Metal carbonyl complexes received more attention due to their potential catalytic and medicinal applications [1-5]. It is also noted that metal carbonyls can easily coordinate with 1,1-bis(diphenylphosphino)methane (dppm) and different other organic reagents [6]. The reactivity of metal carbonyls leads to the formation of a wide variety of metal carbonyl complexes (e.g. [M(dppm)(CO)n]). So, understanding the bonding and electronic structures [7-11] of metal carbonyls is very important as it helps assess the suitability of metal carbonyls as a catalyst or in other fields of application. Additionally, heterobimetallic complexes offer better catalytic features and the synergistic effect provides a unique reactivity feature that is inaccessible by the homobimetallic systems [12]. As we see heterobimetallic complexes and their substitution reactions have been studied extensively using wide classes of ligands e.g., alkynes, phosphines, and phosphites, heterobimetallic complexes are also investigated for sulfur donor ligands. When catalytic processes are studied, sulfur acts as a catalyst poison [13]. It is also noted that along with bimetallic systems metal clusters are also being studied by researchers all over the world over a very long period. Research in heterobimetallic clusters is stimulated by a belief that a combination of metals having diverse chemical and electronic properties may induce unique and highly selective chemical transformations [14]. As pointed out by Geoffroy and co-workers [15], the metal-metal bond (M-M) in heterobimetallic or multi-metallic systems is not purely covalent but can have a significant donor-acceptor character. Such bonds have shown to be relatively weak, being readily cleaved by nucleophiles. It is found that sulfur-containing ligands can be used as precursors for the synthesis of high nuclearity clusters. This happens as the metal sulfur bonds can coordinate with metals using its three available electrons. For example, it was reported the dinuclear complex [CpMoMn(CO)3(µ-CO)(µ-pyS)2] [16] from the thermal reaction of [M2(CO)6(µ-pyS)2] with [CpMo(CO)3]2-. Heteronuclear complexes [CpMoMn(CO)5(µ-E2)] (E=S, Se) were reported by Adams and coworkers [17] from the reactions of [CpMoMn(CO)8] with elemental sulfur and selenium. In the early nineties, Deeming et al. prepared a series of rhenium-ruthenium clusters containing ReRu3 and Re2Ru2 cores in a rather simple way from the reaction of [Re2(CO)6(µ-pyS)2] with Ru3(CO)12 in refluxing xylene, thereby establishing that the dinuclear [Re2(CO)6(µ-pyS)] is an excellent source for the incorporation of an [Re(CO)3(pyS)] unit into the trimetallic system [18]. Here, we are going to report new bimetallic complexes [FeRe(CO)7(κ2-pyS)(κ2-dppm)] (3), [FeRe(CO)7(κ2-pymS)(κ2-dppm)] (5) and known trinuclear [(µ-H)Os3(CO)8(µ-Br)(µ-dppm)] (7) carbonyl complex having different types of coordinating ligands. Complex 7 was reported previously by S. E. Kabir et al., [19] here we are reporting the molecular structure that was not reported.
2. Results and Discussion
- The reaction of [Fe2(CO)8(κ2-dppm)] (1) and [Re2(CO)6(µ-κ3-pyS)2] (2) resulting in the formation of a new complex [FeRe(CO)7(κ2-pyS)(κ2-dppm)] (3) greenish-yellow with 14.40% yield (Scheme 1). The solid-state molecular structure of complex 3 is shown in Figure 1. The molecule contains two different metal centers hold by a dppm ligand. Four carbonyl ligands complete the coordination sphere of the iron atom while three carbonyls and a chelating pyridine-2-thiolato ligand complete the coordination sphere of the rhenium atom.
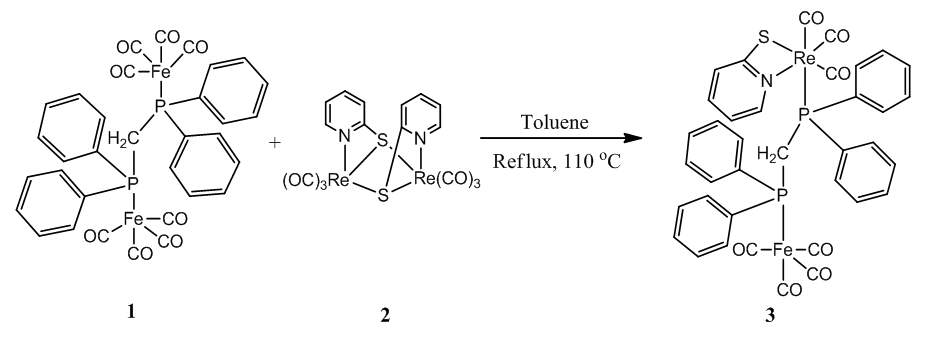 | Scheme 1. Synthesis of the complex [FeRe(CO)7(κ2-pyS)(κ2-dppm)] (3) |
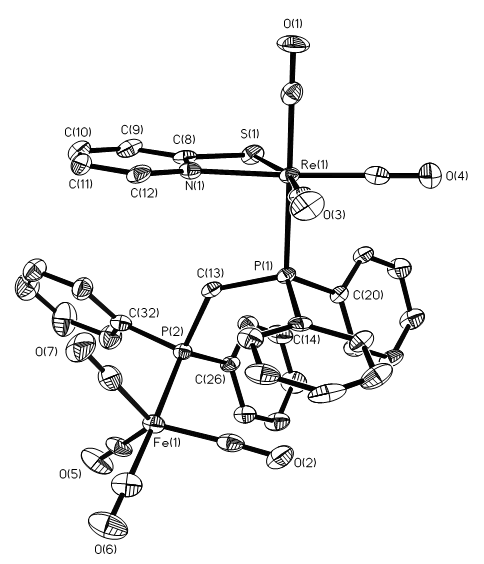 | Figure 1. Molecular structure of [FeRe(CO)7(κ2-pyS)(κ2-dppm)] (3) |
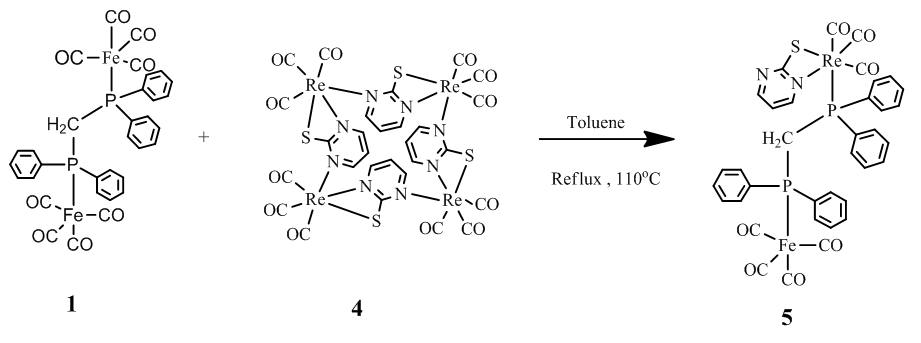 | Scheme 2. Synthesis of the complex [FeRe(CO)7(κ2-pymS)(κ2-dppm)] (5) |
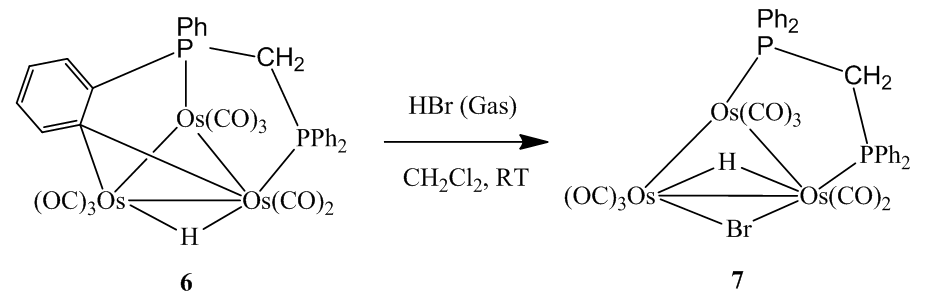 | Scheme 3. Synthesis of the complex [(µ-H)Os3(CO)8(µ-Br)(µ-dppm)] (7) |
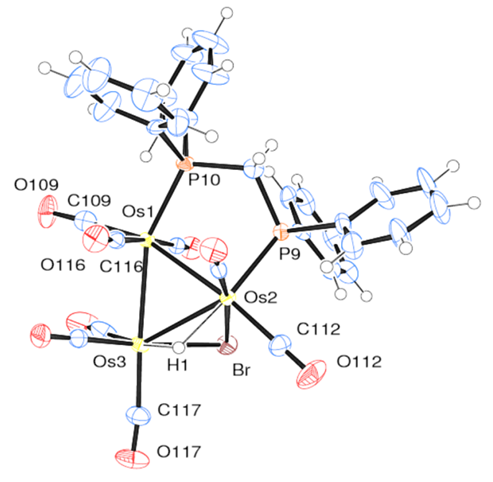 | Figure 2. The solid-state molecular structure of [(µ-H)Os3(CO)8(µ-Br)(µ-dppm)] (7) |
3. Experimental
- Unless otherwise noted, all the reactions were carried out under a nitrogen atmosphere using the standard Schlenk line technique. Solvents were dried using standard drying procedures. Infrared spectra were recorded on a Shimadzu FTIR spectrometer. NMR spectra were recorded on a Bruker DPX 400 instrument. The compounds [Mn2(CO)10], [Fe2(CO)9], [Os3(CO)12], and 1,1-bis(diphenylphosphino)methane (dppm) were purchased from Stream Chemicals Inc. and used as received. The complexes [Fe2(CO)8(κ2-dppm)] [21], [Re2(CO)6(µ-κ3-pyS)2] [22] and [Re4(CO)12(µ-κ3- pymS)4] [23] were synthesized according to literature method.
3.1. Synthesis of the Complex [FeRe(CO)7(κ2-pyS)(κ2-dppm)] (3)
- 0.0370 g (0.067 mmol) of the complex [Fe2(CO)8(κ2-dppm)] and 0.0400 g (0.053 mmol) of the complex [Re2(CO)6(µ-κ3-pyS)2] (40 mg, 0.053 mmol) were stirred at 110oC temperature for 24 h in 30 mL toluene. The reaction cooled and the solution color turned greenish-yellow. The solvent was removed using a rotary evaporator. The compound was separated using preparatory TLC. Elution with hexane/CH2Cl2 (7:3 v/v) gave six bands. The major greenish-yellow band was collected and using DCM the complex [FeRe(CO)7(κ2-pyS)(µ-dppm)] (3) was collected. The slow evaporation and crystallization (DCM/Hexane) in freeze (4°C) gave 9 mg greenish yellow solid (14.40% yield) (Scheme 1). The contents in the other minor bands were too small for complete characterization. IR (νCO, CH2Cl2): 2049 s, 2024 vs, 1975 vw, 1932 (br,vs) and 1900 w cm-1. 1H NMR (CDCl3, 400 MHz): δ 6.5 (t, 1H), 6.6 (d, 1H), δ 7.5 (m, 1H) and 7-7.4 (m, 20H).
3.2. Synthesis of the Complex [FeRe(CO)7(κ2-pymS)(κ2-dppm)] (5)
- 0.0250 g (0.045 mmol) of the complex [Fe2(CO)8(κ2-dppm)] and 0.0290 g (0.0190 mmol) of the complex [Re4(CO)12(µ-κ3-pymS)4] were stirred at 110°C temperature for 47 min in toluene. The solvent was removed using a rotary evaporator, the residue was chromatographed using preparatory TLC, and using the solvents hexane/CH2Cl2 (5:5 v/v) gave two bands. The major band gave the bimetallic complex [FeRe(CO)7(κ2-pymS)(µ-dppm)] (5) (12mg, 28.39%) as pale yellow crystals after recrystallizing from hexane/CH2Cl2 in a freeze at 4°C temperature (Scheme 2). Spectral data for 5: IR (νCO, CH2Cl2): 2050 s 2027 vs, 1976 w, 1936 (br,vs), 1903 w cm-1.
3.3. Synthesis of the Complex [(µ-H)Os3(CO)8(µ-Br)(µ-dppm)] (7)
- 0.125 g (0.106 mmol) of the complex [(µ-H)Os3(CO)8(Ph2PCH2P(Ph)C6H4)] (6) was dissolved in 35 mL of CH2Cl2 solution. HBr gas was bubbled into the solution at room temperature for 2 min during which time the color was changed from green to red (Scheme 3). The solvent was then removed under reduced pressure and the residue was chromatographed using preparatory TLC. Eluting with solvents hexane/CH2Cl2 (1:1, v/v) gave one orange band which afforded [(µ-H)Os3(CO)8(µ-Br)(µ-dppm)] (7) (0.127 g, 95%) as red crystals after recrystallization from hexane/CH2Cl2 at -4°C putting in a freeze. (The spectroscopic characterization of the complex was reported by S. E. Kabir et al., [19]. Here we are reporting XRD structure).
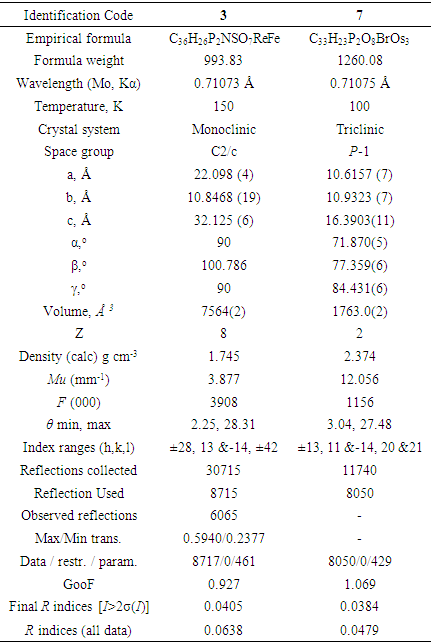
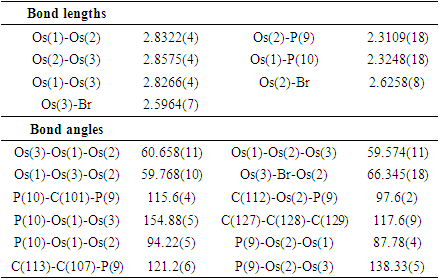
3.4. X-ray Structure Determinations
- Single crystals were mounted on fibers and diffraction data was collected at low temperature (see Table 1) on Bruker AXS SMART APEX II CCDD diffractometer using Mo-Kα radiation (λ=0.71073Ao). Data collection, indexing, and initial cell refinements were all done using SMART Apex-II [24] software. Data reduction was accomplished with SAINT software [25] and the SADABS program [26] was used to apply empirical absorption corrections. The structures were solved by direct method [27] and refined by full-matrix least-squares [27]. All non-hydrogen atoms were refined anisotropically and hydrogen atoms were included using a riding model. Scattering factors were taken from International Tables for X-ray Crystallography [28]. Additional details of data collection and structure refinement are given in Table 1.
|
|
4. Conclusions
- The bimetallic complexes [FeRe(CO)7(κ2-pyS)(κ2-dppm)] (3) and [FeRe(CO)7(κ2-pymS)(κ2-dppm)] (5) have been synthesized. The structures of the complexes [FeRe(CO)7(κ2-pyS)(κ2-dppm)] 3 and [(µ-H)Os3(CO)8(µ-Br)(µ-dppm)] (7) are also confirmed from XRD studies. Finally, the synthesis of the complexes is opening a new route for synthesizing new bimetallic and trimetallic carbonyl complexes of different transition metals and exploring their applications.